|
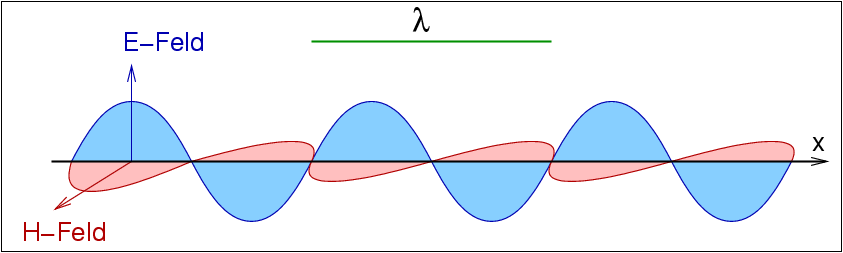
| Abb. I.1.1. Charakteristika elektromagnetischer Strahlung ‣ SVG |
| cr_home | Metalle | Nichtmetalle | FK-Chemie | Strukturchemie | Interm. Phasen | Oxide | Silicate | Strukturtypen | AFP |
| ← | Inhalt | Einleitung | I. Spektroskopie | II. Beugung | III. Bildgebung | IV. Sonstige Methoden | → |
Vorlage(n)
|
|
| Abb. I.1.1. Charakteristika elektromagnetischer Strahlung ‣ SVG |
|
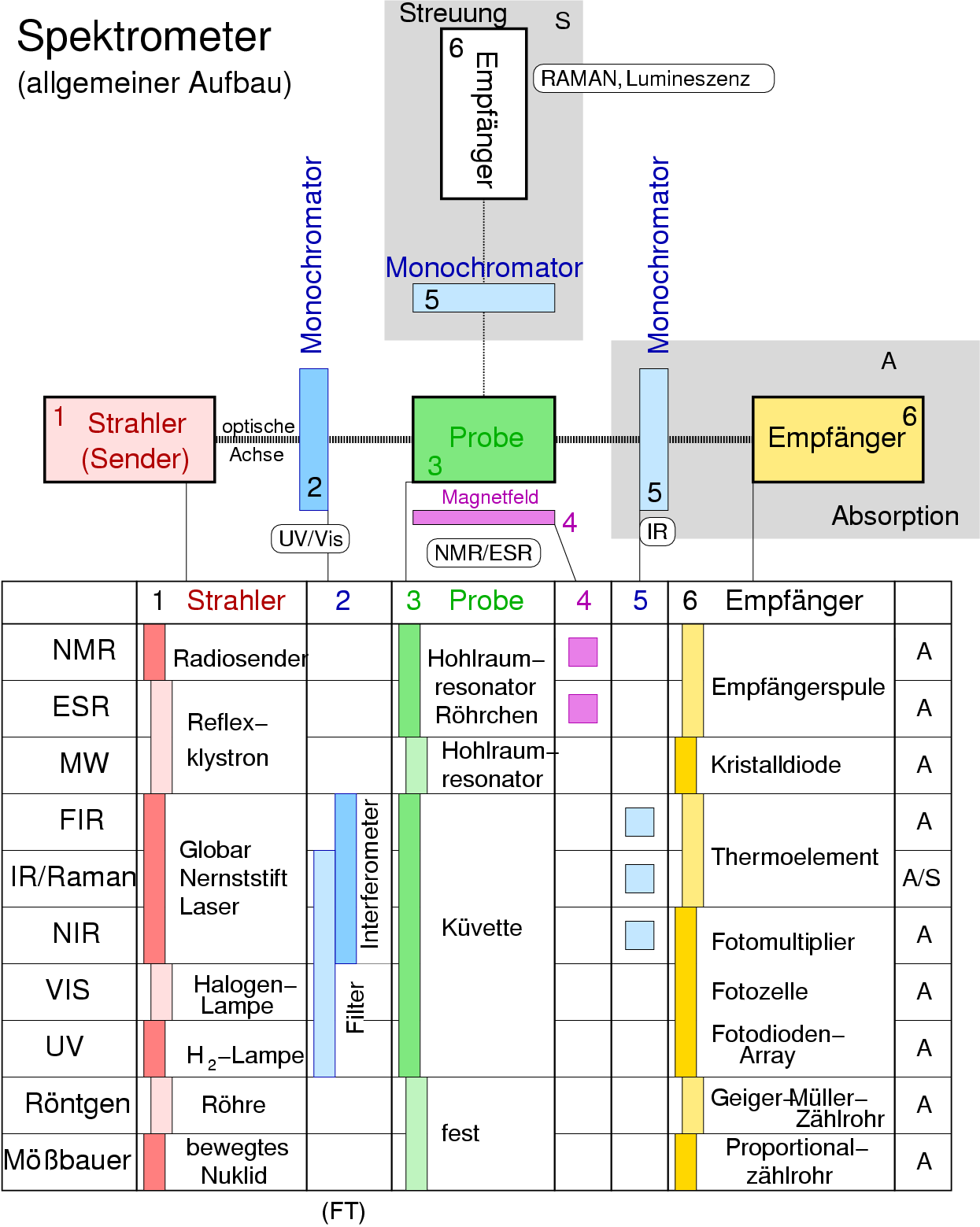
| Abb. I.1.2. Spektrometer (allgemein, schematisch) ‣ SVG |
|
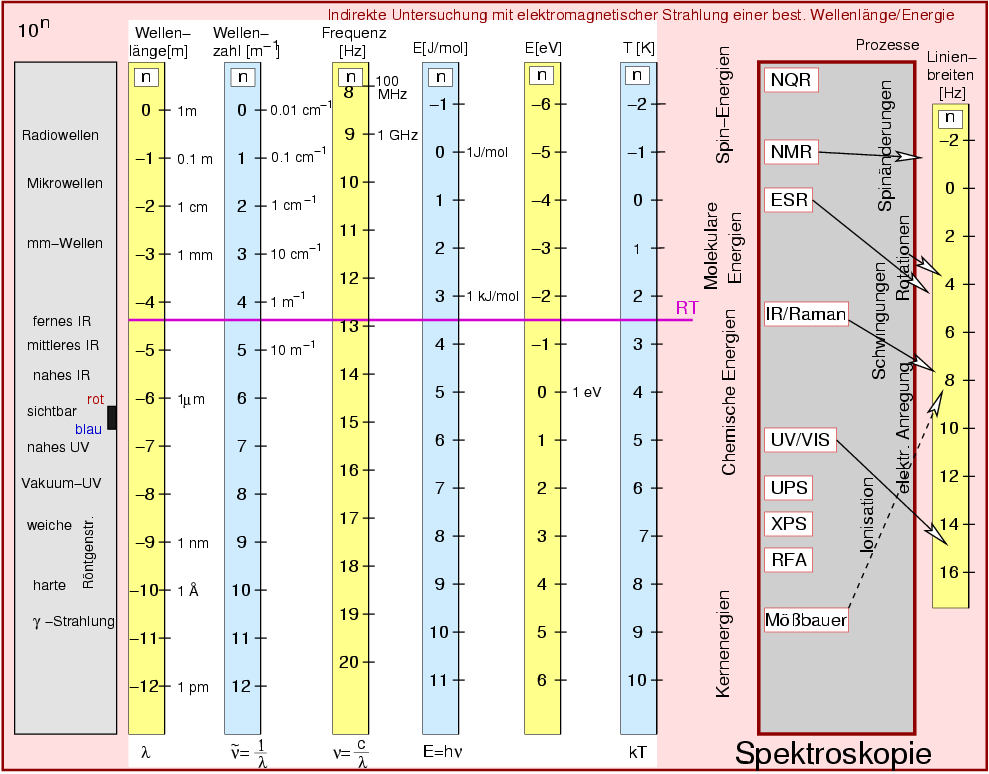
| Abb. I.1.4. Energien und Linienbreiten bei der Spektroskopie ‣ SVG |
|
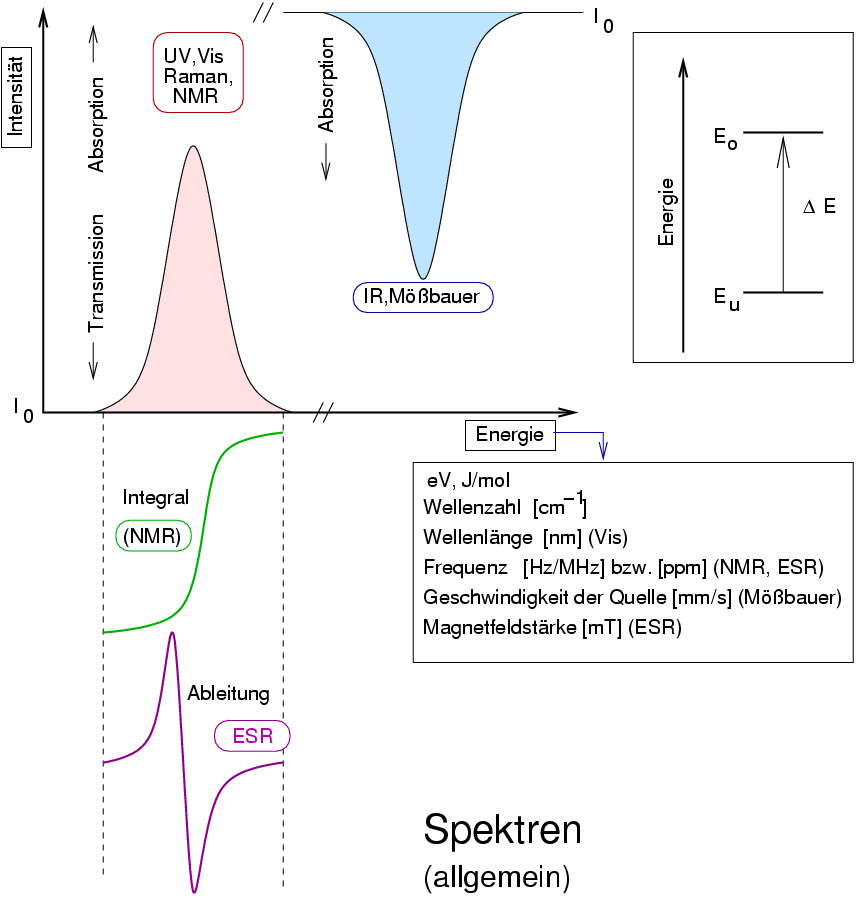
| Abb. I.1.3. Spektrum (allgemein, schematisch) ‣ SVG |
Beispiele
Beispiele:
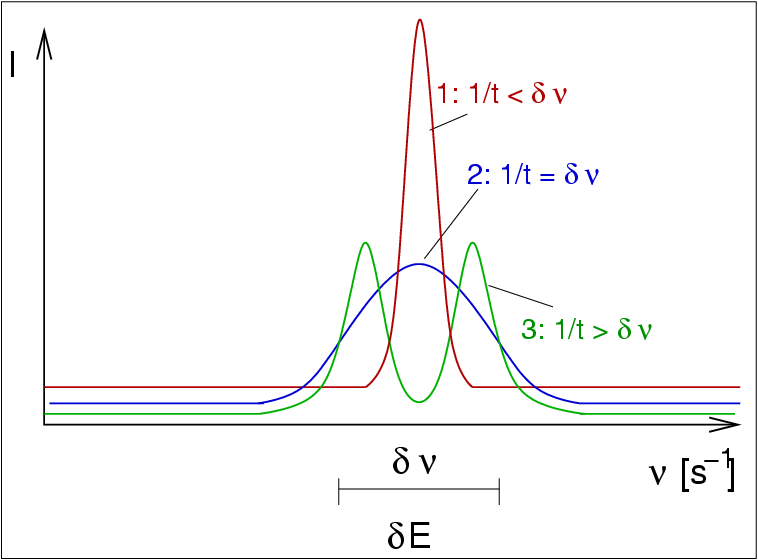
|
| Abb. I.1.5. Linienbreiten und dynamische Prozesse in der NMR-Spektroskopie ‣ SVG |
| ← | Inhalt | Einleitung | I. Spektroskopie | II. Beugung | III. Bildgebung | IV. Sonstige Methoden | → |
| cr_home | Metalle | Nichtmetalle | FK-Chemie | Strukturchemie | Interm. Phasen | Oxide | Silicate | Strukturtypen | AFP |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}