Elektronen sind (vgl. UV/VIS-Spektroskopie) Fermion mit einer Spinquantenzahl von s=1/2. Der Betrag des Spins bzw. des mechanischen Eigendrehimpulses zeigt eine Betragsquantelung: Die Orientierungsquantenzahl (ms), die die Orientierung im Magnetfeld beschreibt, kann nur die Werte ms = -s, ... +s, d.h. ± 1/2 annehmen. Daraus ergeben sich 2s+1=2 Einstellungen im Feld, die ohne angelegtes Feld entartet sind. Die z-Komponenten des Elektronenspins (Richtungsquantelung) kann nur Werte von (Richtungsquantelung) annehmen. Das rotierende geladene Teilchen ist ein magnetischer Dipol (Minimagnet) mit einem Dipolmoment in [J/T] wobei βB das Bohrsche-Magneton und γ das gyromagnetische Verhältnis bedeutet.
Isolierte Nukleonen (Protonen p und Neutronen n) alleine sind ebenfalls Fermion mit einem Einzeldrehimpuls von s=1/2. Die Gesamtkerne zeigen einen Kernspin (mit Spinquantenzahl I), der durch Vektoraddition von p- und n-Spin (wie genau ist nur durch die aktuelle Kernphysik erklärbar) zustande kommt. Hierfür gelten jedoch folgende einfache Regeln:
| Kernspin-Quantenzahl | halbzahlig | 0 | ganzzahlig |
|---|---|---|---|
| Massenzahl | u | g | g |
| Kernladungszahl | g oder u | g | u |
| Isotop | Kernspin | Häufigkeit [%] | γ [107 rad T-1s-1] | rel. Frequenz [MHz] | rel. Empfindlichkeit |
|---|---|---|---|---|---|
| 1H | 1/2 | 99.985 | 26.7519 | 100 | 1.0 |
| 2H | 1 | 0.015 | 4.1066 | 14.7 | 1.5 . 10-6 |
| 10B | 3 | 19.6 | 2.8746 | 10.7 | 3.9 . 10-3 |
| 11B | 3/2 | 80.4 | 8.5843 | 32.1 | 1.3 . 10-1 |
| 13C | 1/2 | 1.11 | 6.7283 | 76.2 | 5.8 . 10-7 |
| 14N | 1 | 99.6 | 1.9338 | 7.2 | 1.0 . 10-3 |
| 15N | 1/2 | 0.37 | -2.712 | 10.1 | 3.9 . 10-6 |
| 19F | 1/2 | 100.0 | 25.181 | 94.1 | 8.3 . 10-1 |
| 27Al | 5/2 | 100.0 | 6.9760 | 26.1 | 2.1 . 10-1 |
| 31P | 1/2 | 100.0 | 10.841 | 40.5 | 6.6 . 10-2 |
| 73Ge | 9/2 | 7.8 | -0.9357 | 3.5 | 1.1 . 10-4 |
| 197Au | 3/2 | 100 | 4.2342 | 16.2 | 51.4 . 10-1 |
- 1H mit I=1/2: 2 Einstellungen
- 14N mit I=1: 3 Einstellungen (s. Abb. I.3.1.)
- 11B mit I=3/2: 4 Einstellungen und bei
- 73Ge mit I=9/2 sogar 10 Einstellungen
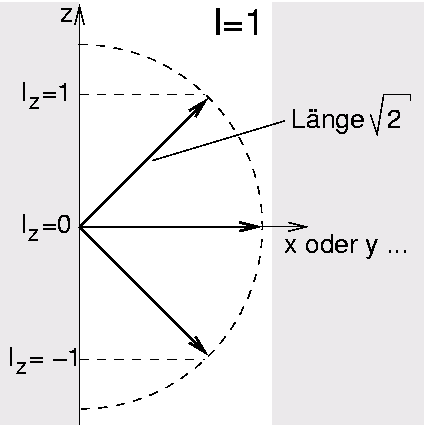
|
| Abb. I.3.1. Drehimpuls und Iz für Kerne mit I=1 ‣ SVG |
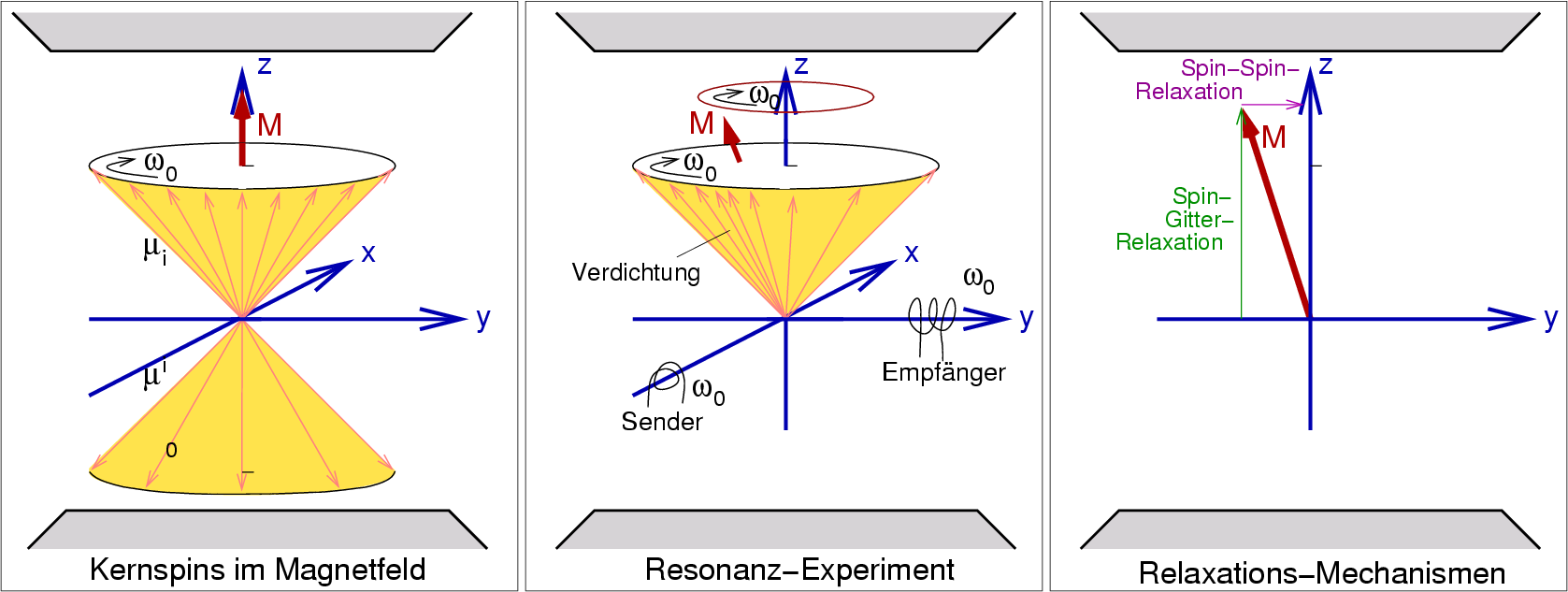
|
| Abb. I.3.2. Funktions-Prinzip der NMR-Spektroskopie ‣ SVG |
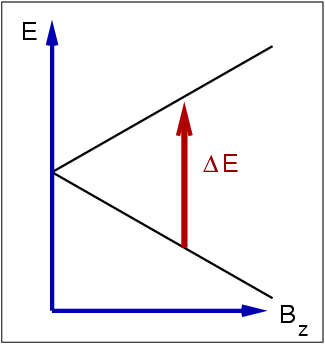
|
| Abb. I.3.3. Abhängigkeit von ΔE vom Magnetfeld Bz ‣ SVG |
- 1H bei 2.3 T: 100 MHz
- 13C bei 2.3 T: 25 MHz
- Früher hat man im Frequenzabtastverfahren (sog. CW = Continuous Wave) ν mit der Senderspule durchgefahren. Wenn die Frequenz der eingestrahlten Strahlung der Lamorfrequenz entspricht, kommt es zum Umklappen der Spins (Spinflip), die Strahlung wird absorbiert. Der Vektor M verbleibt in z-Richtung, ändert jedoch sein Länge. In der Empfänger-Spule, die um die Probe herum angeordnet ist und senkrecht zur Senderspule liegt, wird ein Induktionsstrom induziert. Diese Methode ist sehr langsam, da die Frequenzen langsam durchgefahren werden müssen.
- Heute wird als Standard-Verfahren 1D-Fourier-Transform-NMR (FT-NMR) verwendet.
Ein kurzer (wenige Mikrosekunden), nur wenige Grad des Lamor-Umlaufs langer 90o-Frequenzpuls, der alle
Frequenzen zugleich enthält und ein kleines Magnetfeld in dieser Richtung aufweist,
bewirkt die Ausrichtung der energetisch passenden Gesamtmagnetisierung M
in die x-y-Ebene. Die Spins werden auf dem Kegelmantel verdichtet (Abb. I.3.2. Mitte, hier sind
nur noch die Nichtgleichgewichts-Spins dargestellt!), die Gesamtmagnetisierung
gegen die z-Richtung des Magneten ausgelenkt (Quermagnetisierung).
Nach dieser Einstrahlung kehrt die Gesamtmagnetisierung (aufgrund verschiedener Relaxationsprozesse, s.u.)
mit der Lamor-Frequenz in den
Gleichgewichtszustand zurück, in der Empfängerspule (in y-Richtung) wird das mit der Zeit exponentiell
abfallende Signal registriert (sog. FID = free induction decay).
Die Pulse werden ca. alle 3-5 s wiederholt, und die
Spektren akkumuliert. Die Fourier-Transformation des Gesamt-FIDs (Zeitspektrum) ergibt dann das bekannte
Frequenz-Spektrum (s. Abb.I.3.4).
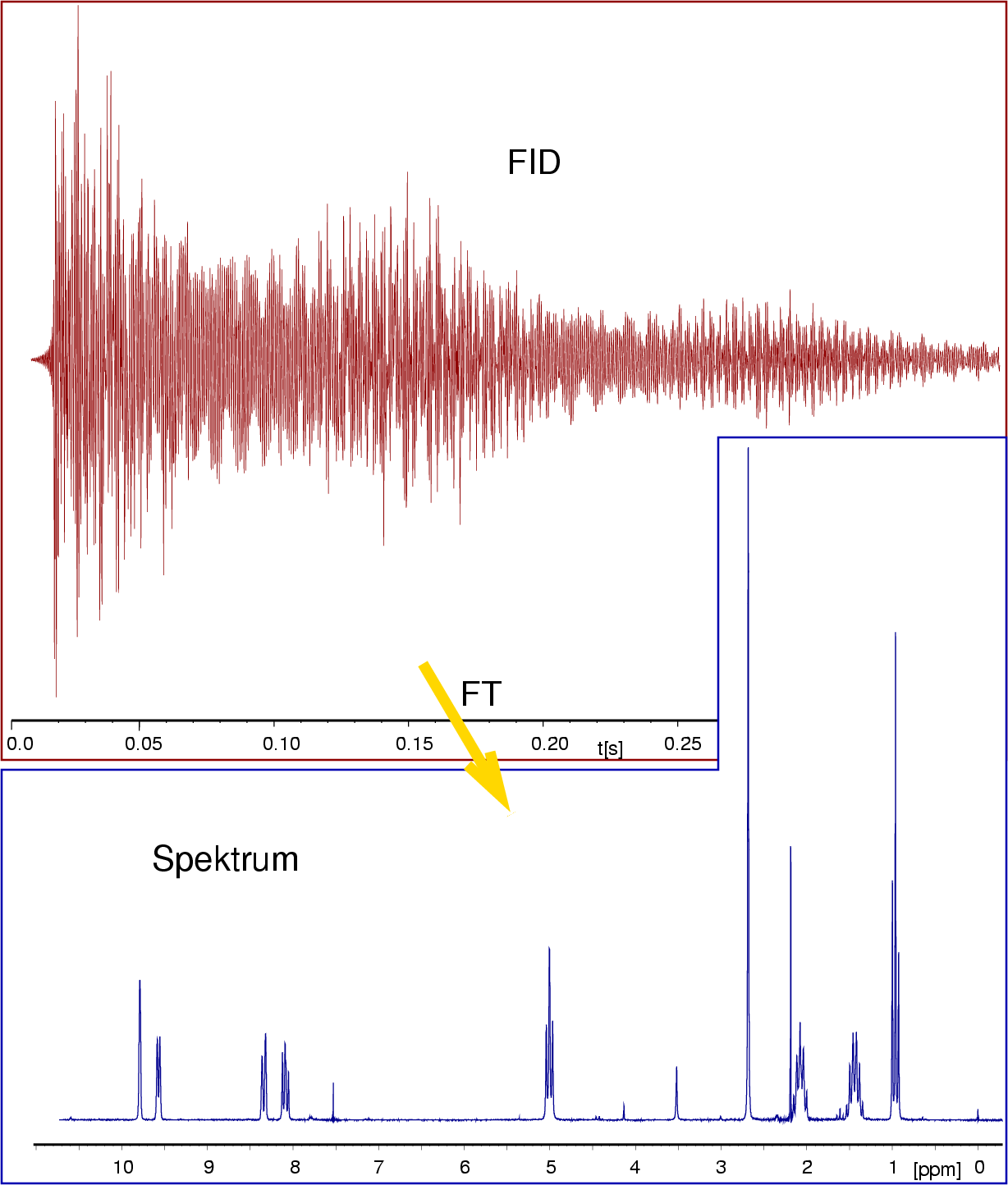
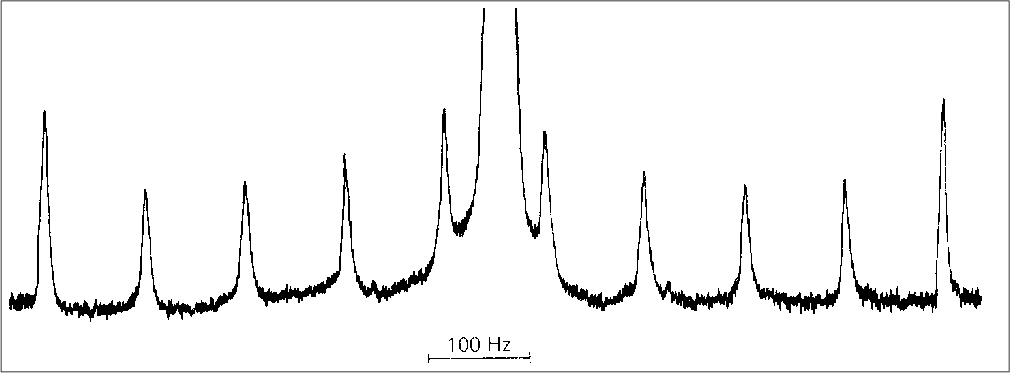
Abb. I.3.4. FID (rot) und das durch Fourier-Transformation erhaltene Spektrum (blau) ‣ SVG - Bei modernen Mehrdimensionalen-NMR-Techniken werden meist komplizierte Pulsfolgen (Spin-Echo-Experiment) gefahren (2D heteronukleare NMR (COSY=COrrelated SpectroscopY). Einige auf diese Weise erhaltene Spektren werden unten bei den Beispielen gezeigt, die Details dieser Messungverfahren überlassen wir der PC.
Nach der Störung des Gleichgewichts kehren die Spins durch verschiedene Relaxations-Prozesse in das Boltzmann-Gleichgewicht zurück. Je nach Messverfahren können die einzelnen Relaxationsprozesse auch einzeln vermessen werden. Man unterscheidet in (s. Abb. I.3.2. rechts):
- Bei der Spin-Gitter- oder longitudinalen Relaxation oder T1 erfolgt ein Austausch mit der Umgebung (dem Gitter), in dem immer passende Frequenzen vorhanden sind. Der energetisch höhere Zustand wird wieder in den tieferliegenden umgewandelt. Die typischen Zeiten für diese Prozesse, die verglichen mit der Lamor-Frequenz extrem groß sind, liegen bei Festkörpern bei 10-2 bis 104 s, d.h. die Gesamtmagnetisierung wird nur sehr langsam wieder aufgebaut. Die Zeiten liegen bei flüssigen Proben zwischen 10-4 und 1 s, meist bei ca. 10-2 s.
- Bei der Spin-Spin- oder transversale Relaxation oder T2 kommt es zur Wechselwirkung der 'Ungleichgewichts'-Spins untereinander, dadurch wird die Verdichtung abgebaut. Typische Zeiten sind im Festkörper 10-4 s (sehr schnell!), in flüssigen Proben typischerweise ca. 10-2 s.





















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}