
| cr_home | Metalle | Nichtmetalle | FK-Chemie | Methoden | Interm. Phasen | Oxide | Silicate | Strukturtypen |
| ⇦ | Inhalt | Einleitung | Kovalente FK | Metalle | Ionenkristalle | Literatur | ⇨ |
Die Bindigkeit der Atome in den Elementen der 6. Hauptgruppe ist nach der 8-N-Regel zwei. Die Strukturchemie wird entsprechend von Ketten oder Ringen, d.h. 0-dimensionalen bzw. 1-dimensional unendlichen Bauverbänden geprägt. Das erste Element der Gruppe, der Sauerstoff, geht auch Mehrfachbindungen ein und bildet entsprechend hantelförmige Moleküle mit einer formalen Doppelbindung (Diradikal, s. MO-Schemata aus der Vorlesung Chemie der Nichtmetalle, darin Kap. 5.1., Abb. 5.1.3 sowie VRMLs der einzelnen MOs).
| flüssig | < 54 K | γ | < 43.8 K | β | < 24 K | α | |
| Dichte | . | . | 1.32 | . | 1.334 | . | 1.495 |
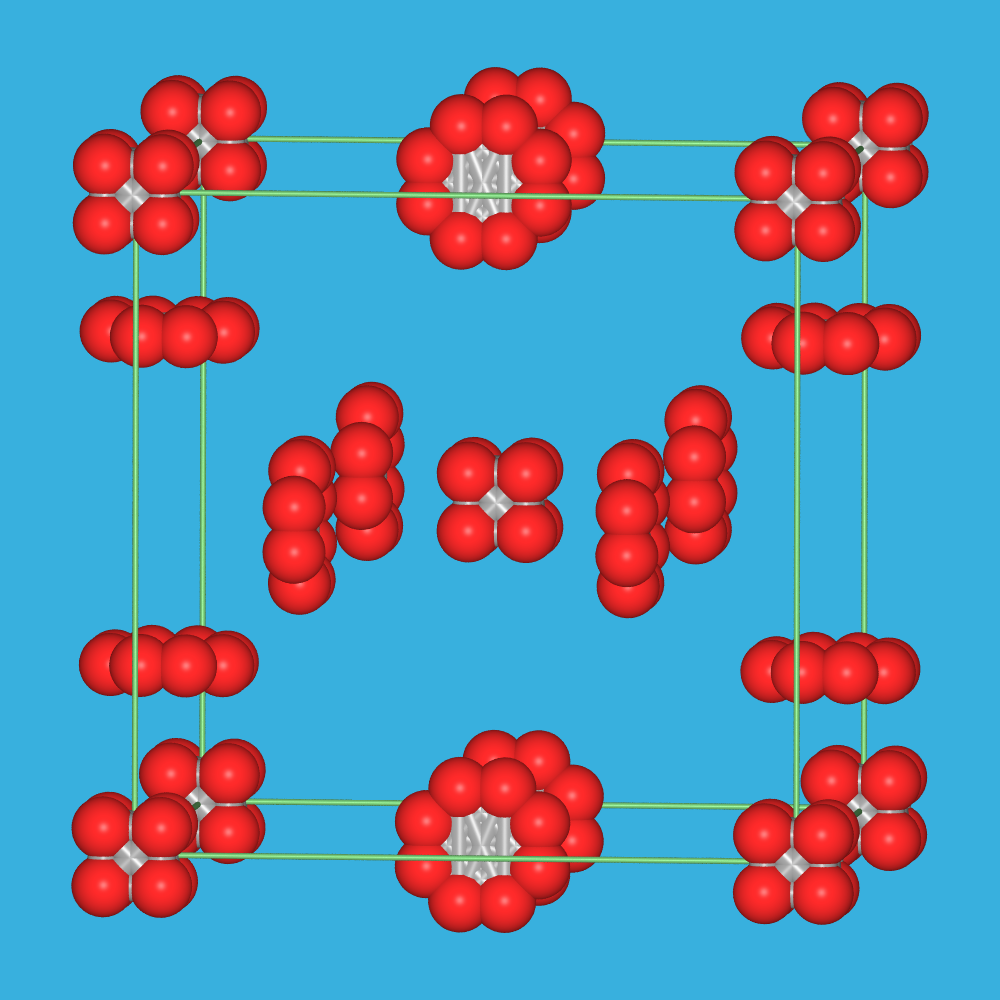
| Struktur | cP, Cr3Si | rhomboedrisch (f.c.c.-analoge Stapelfolge) | monoklin (C2/m) | ||||
| O2 nur partiell fehlgeordnet, β-F2-Typ | O2 entlang [111] nach |:ABC:| ausgerichtet | |:AD:|-Stapelung über Kanten (vgl. α-U-Typ) | |||||
| Elementarzelle |
|
|
| ||||
| VRML | Zelle | Zelle | Zelle |
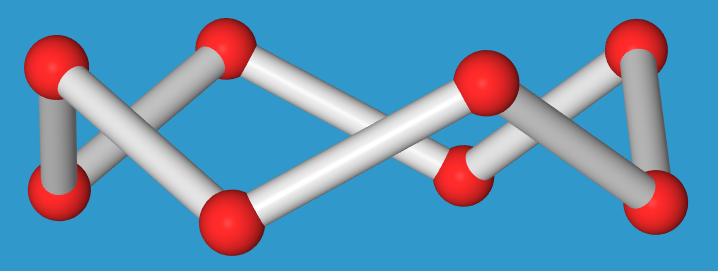
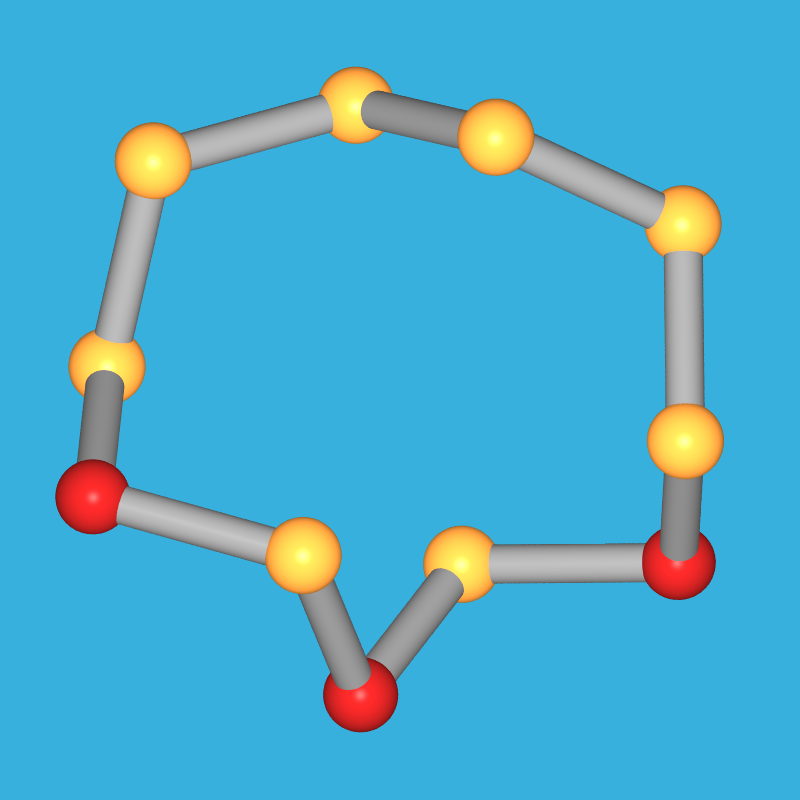
Die Abstände und Winkel liegen in allen Formen im Bereich dS-S von ca. 206 pm (+/- 10 pm), die Winkel am Schwefel zwischen 101 und 110o, was der Voraussage durch die VSEPR-Regeln entspricht. Der Dieder-Winkel (bei vier verknüpften Atomen ABCD Winkel zwischen den Ebenen durch ABC und BCD) liegt im Bereich zwischen 74 und 100 o. Damit ergeben sich Freiheiten in der Gesamtstruktur praktisch erst ab dem fünften Atom. Man unterscheidet dann in cisoide und transoide Verknüpfung (Abb. 2.2.3.2).
Die beiden Verknüpfungsarten treten auf bei:
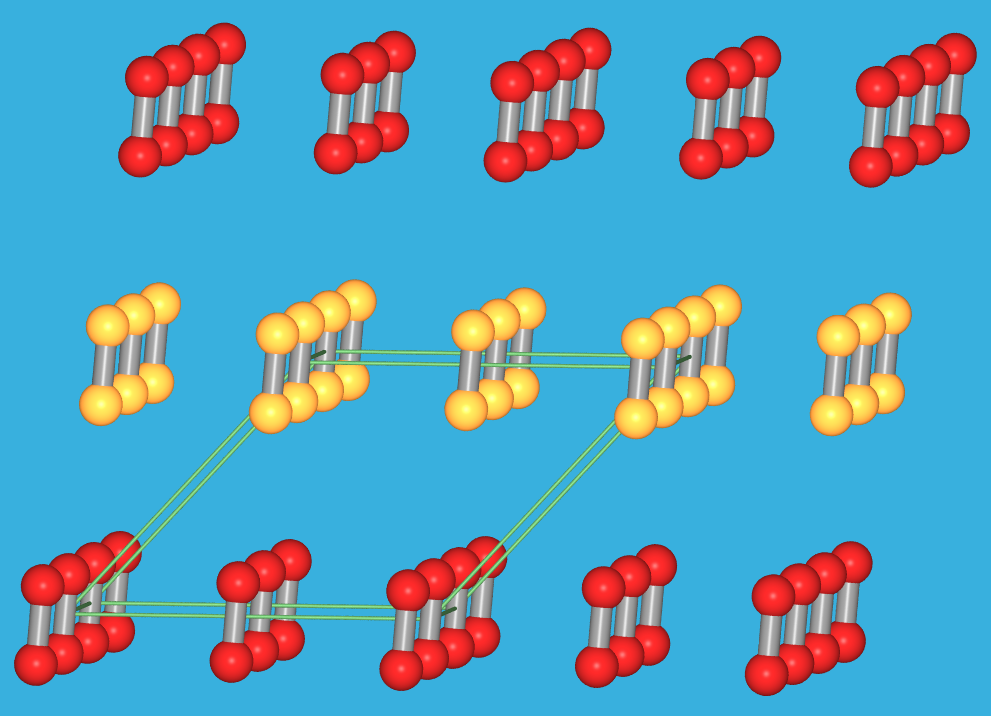
| α-S8 (orthorhombisch) | β-S8 (monoklin) | ||
 |
 |
 |
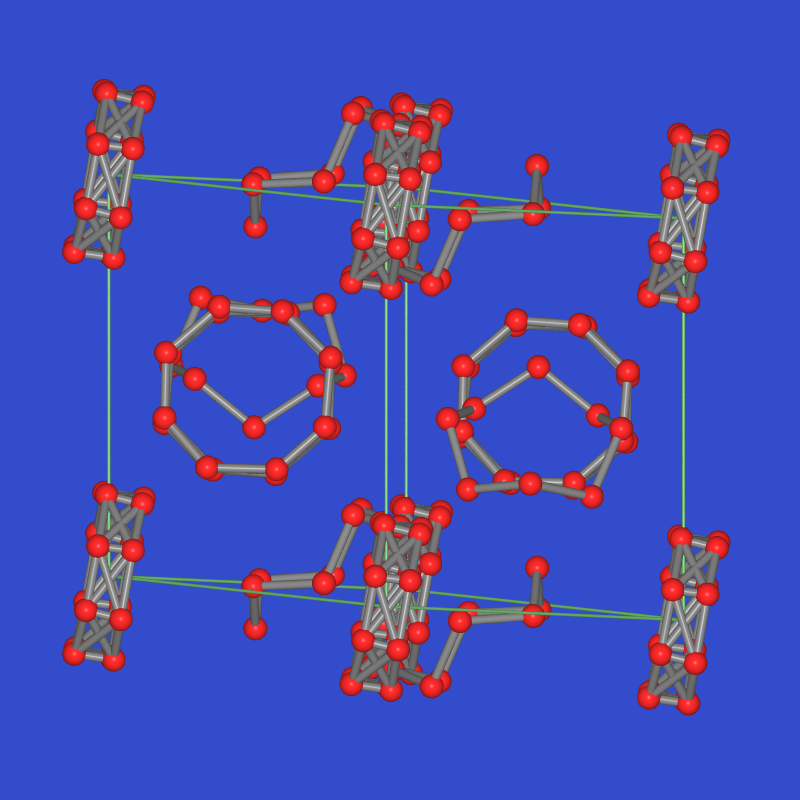 |
| Einzelmoleküle, eine Rolle, Elementarzelle mit markierten Rollen in unterschiedliche Richtungen, (s.a. ST-DB) | Elementarzelle (!! die S8-Ringe sind z.T. fehlgeordnet!) | ||
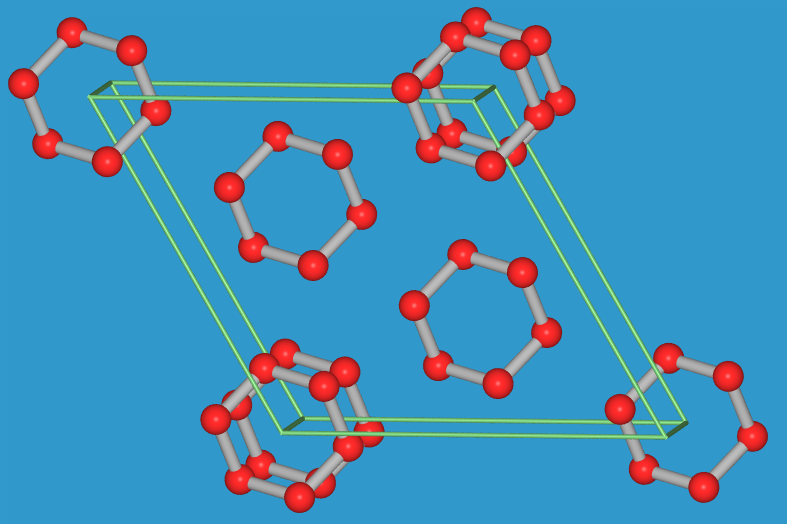
| S6-Ringe | S7-Ringe (γ-Form) | S10-Ringe | S11-Ringe |
 |
 |
 |
 |
| VRML | VRML | VRML | VRML |
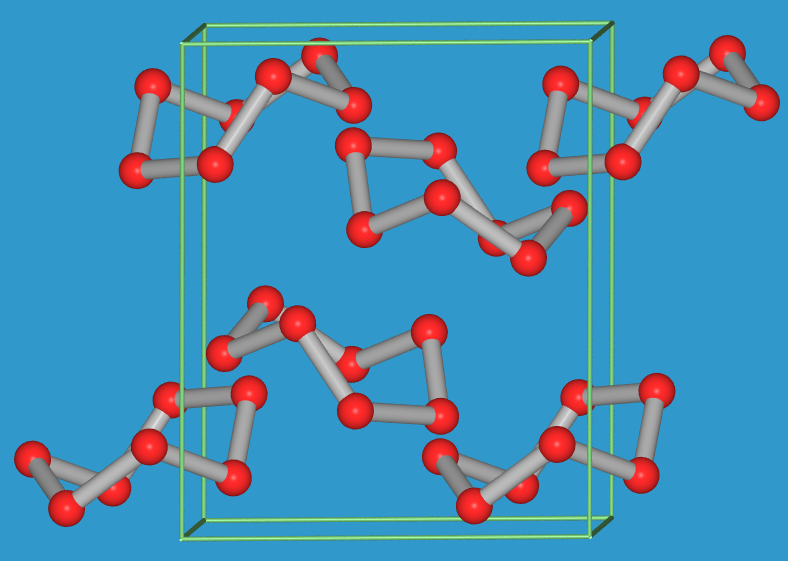
| S12-Ringe | S18-Ringe | S20-Ringe | Sx-Kette |
 |
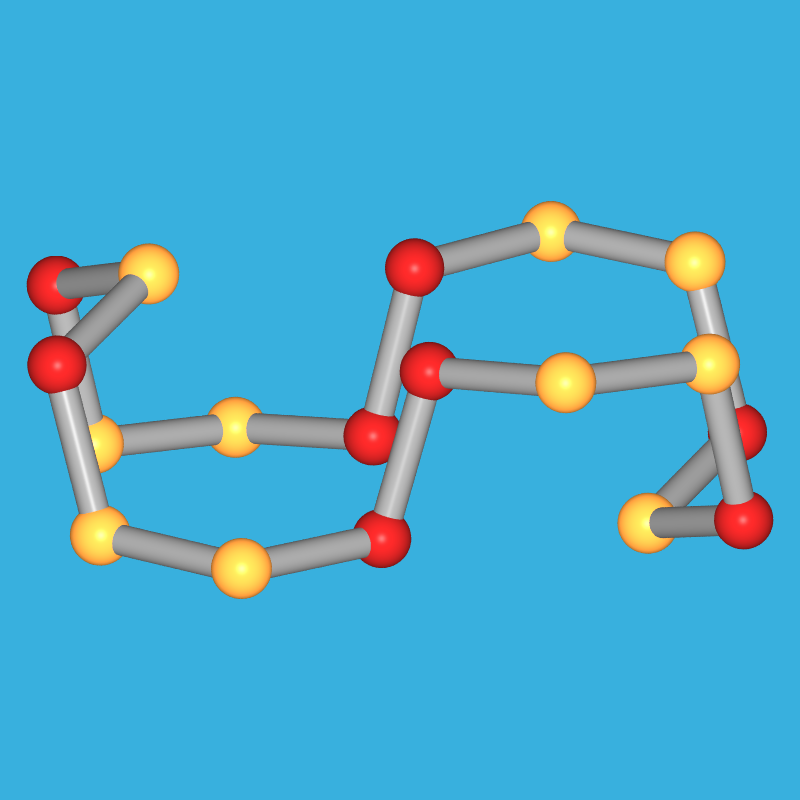 |
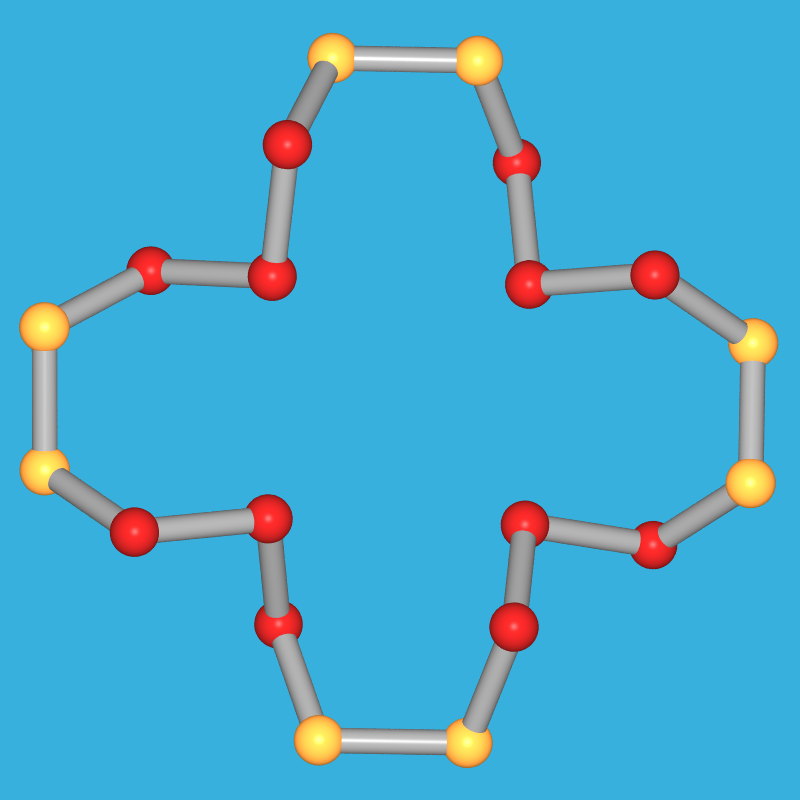 |
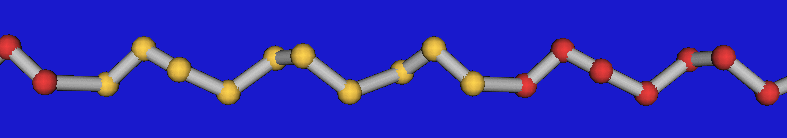 |
| VRML | VRML | S20-Ringe | einzelne Kette |
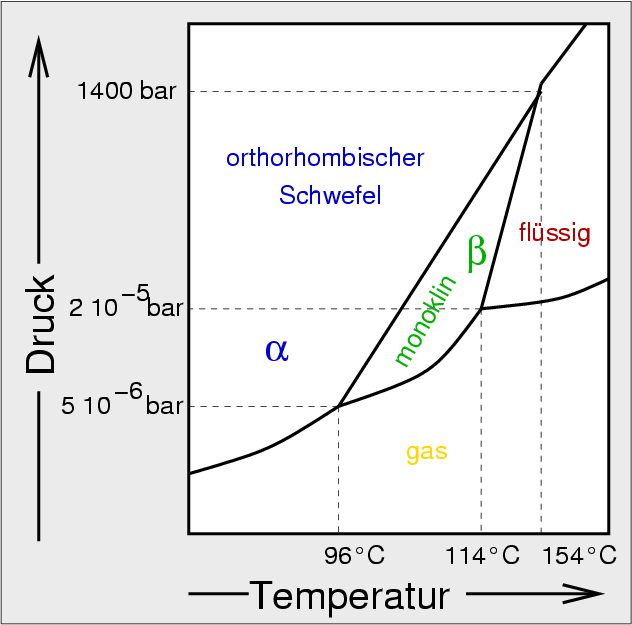
Alle festen thermodynamisch stabilen Formen von Schwefel enthalten S8-Ringe. Die Winkel S-S-S betragen ca. 108o, die S-S-Abstände ca. 204 pm. Abhängig von Druck und Temperatur ist im Feststoff die α- oder die β-S8-Modifikation stabil, wie das Phasendiagramm in Abbildung 2.2.3.4. zeigt.
 |

|
 |
| Abb. 2.2.3.4. p-T-Diagramm von Schwefel ‣SVG | Abb. 2.2.3.5. Natürlicher kristalliner α-Schwefel (links) und aus p-Xylol kristallisierter β-Schwefel (15.11.2019, Dank an C. Serr-Gehring/Versuch und H. Schuster/Foto). | |
| ⚗Kristallisation von monoklinem Schwefel (18MB|MP4|H264) |
Beim Aufheizen von elementarem α-Schwefel bei Normaldruck kommt es zu einer Reihe von Phasenumwandlungen:
| ⚗ Schmelzen und Abschrecken von Schwefel (26MB|MP4|H264) |
Zur Herstellung definierter Ringe Sn sind jeweils spezielle Verfahren nötig, die Abtrennung einzelner Spezies kann auch durch Chromatographie erfolgen.
Kleinere Ringe kommen in S6, das die wohlbekannte Sesselform, d.h. reine cisoide-Konformation, aufweist, und in S7 (mit zwei verschiedenen Modifikationen) vor.
Größere Ringe Sn sind für n = 10, 11, 12, 13, 18 und 20 bekannt, wobei mit steigender Ringgröße immer mehr transoide-Verknüpfung (in den Abb. 2.2.3.3. gelb markiert) beobachtet wird.
In den unendlichen Ketten Sx, die als 103-Schraubenketten kristallisiert werden konnten, kommt nur noch die transoide-Verknüpfung vor.
 |
 |
 |
| Abb. 2.2.3.6. Selen | Abb. 2.2.3.7. Glasiges Selen | Abb. 2.2.3.8. Tellur |
| Sex-Kette | Se-Po | Po |
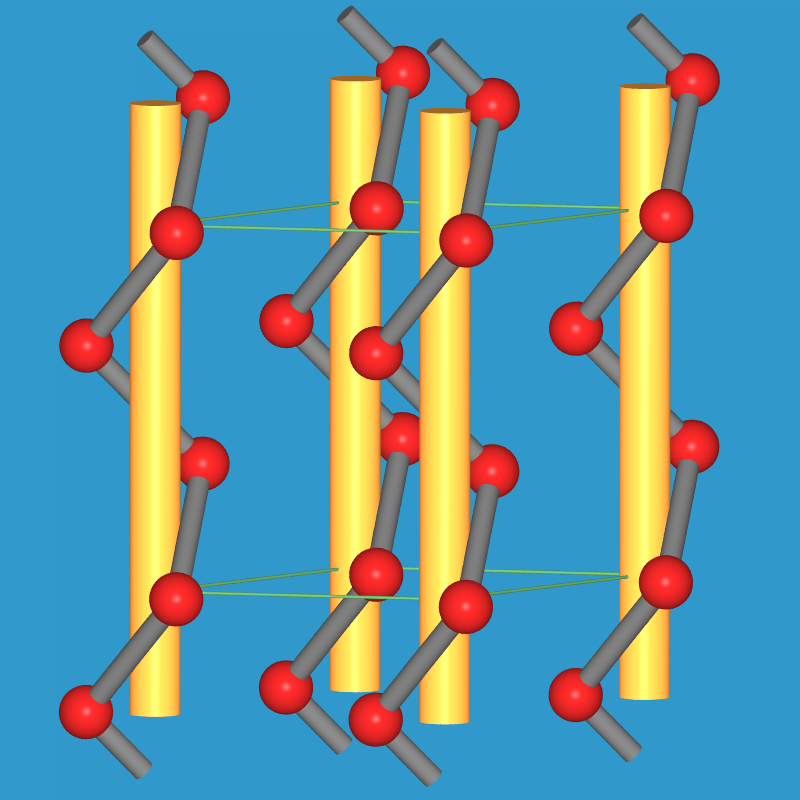 |
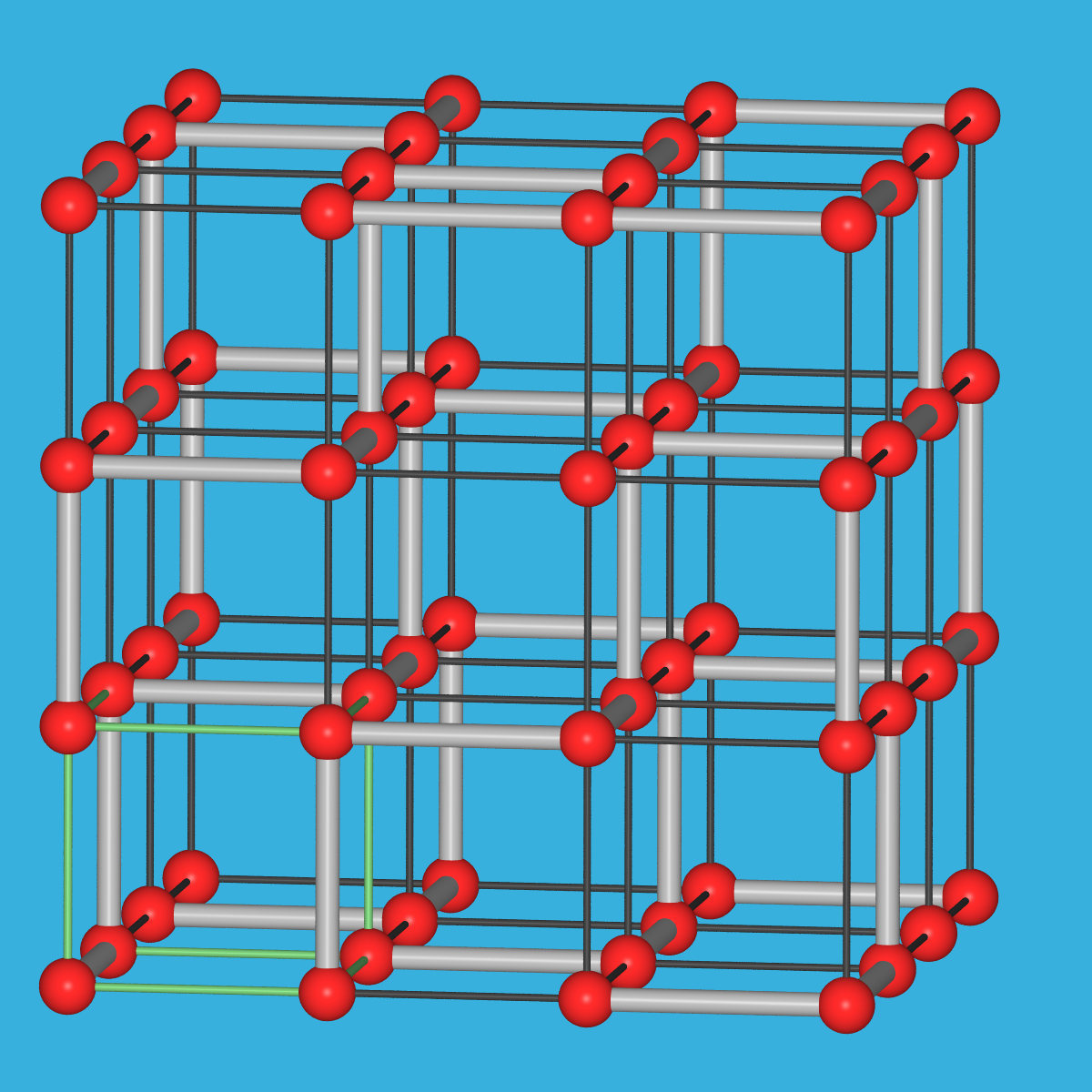 |
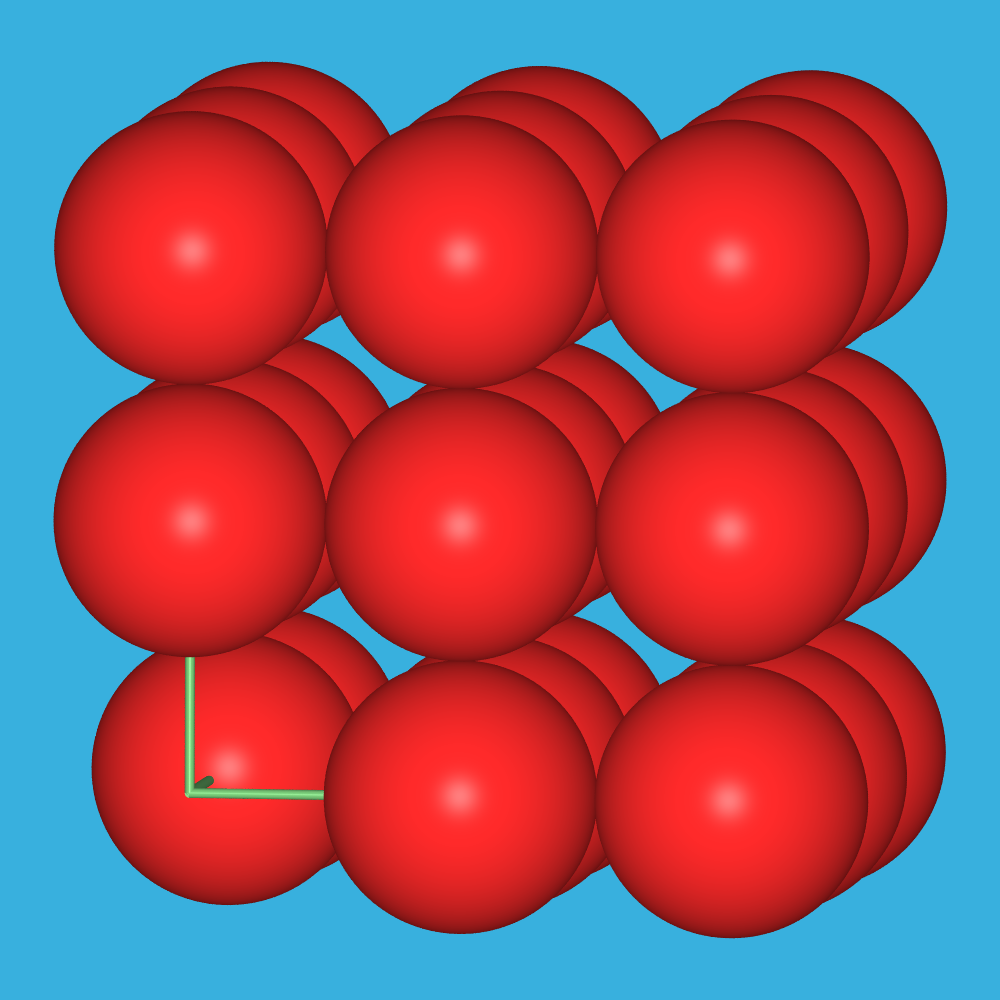 |
| Se-Schraubenketten (s.a. ST-DB) | Strukturverwandtschaft Se-Po | α-Po-Strukturtyp |
Bemerkenswert ist, dass sich die Kristallstruktur von Selen und Tellur aus der
Polonium-Struktur entwickeln läßt, indem die Atome von der regelmäßigen
Oktaeder-Koordination in eine 2+4 Koordination verschoben werden (Peierls-Verzerrung, Abb. 2.2.3.9 Mitte).
In der Abbildung 2.2.3.11. sind die Druckmodifikationen der Elemente
Selen und Tellur (sowie der Pentele, s. folgendes Kapitel 2.2.4.)
grafisch zusammengefasst.
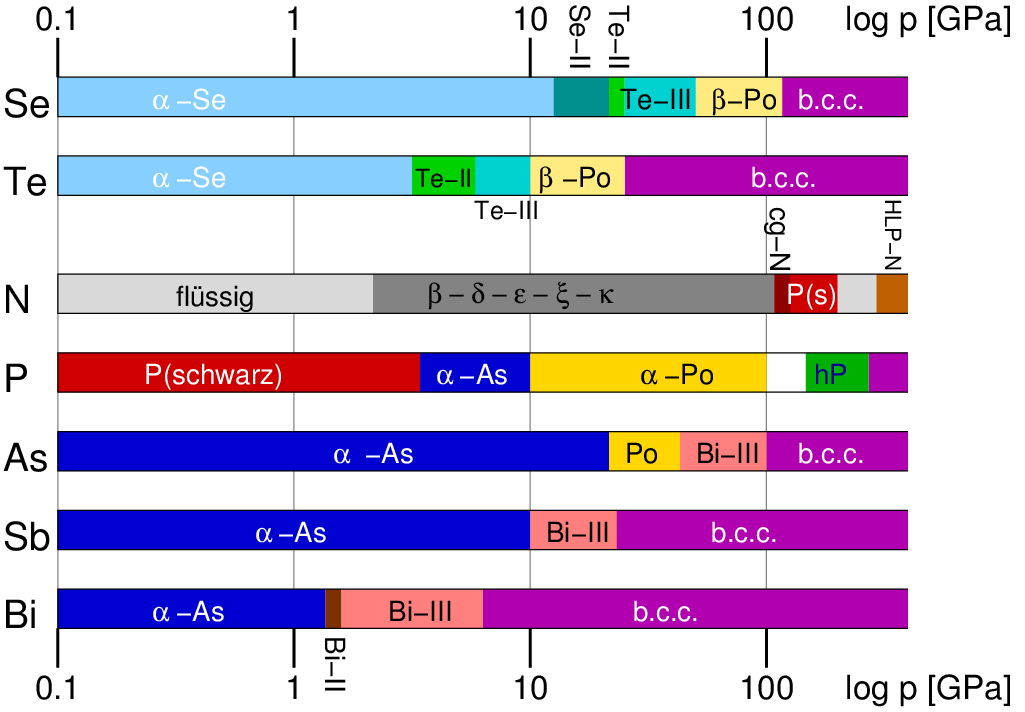 |
| Abb. 2.2.3.10. Druckmodifikationen von Se und Te im Vergleich mit den HP-Formen der Pentele ‣SVG |
Bei höheren Drücken erfolgt über Zwischenstufen wie Se-II, Te-II und Te-III, bei denen sich bereits die Erhöhung der Koordinationszahl auf sechs andeutet (s. die monokline Struktur von Te-II in Abb. 2.2.3.11), die Umwandlung zunächst in die leicht verzerrte Polonium-Struktur (kubisch primitiv, β-Po mit leicht gestauchtem Würfel) mit einer echten Sechsfach-Koordination (Druckhomologen-Regel). Bei weiter erhöhten Drücken von ca. 100 GPa bei Selen und ca. 20 GPa bei Tellur gehen die beiden Elemente schliesslich in den metallischen Wolfram-Typ (b.c.c.), mit einer Koordinationszahl von 8+6, über.
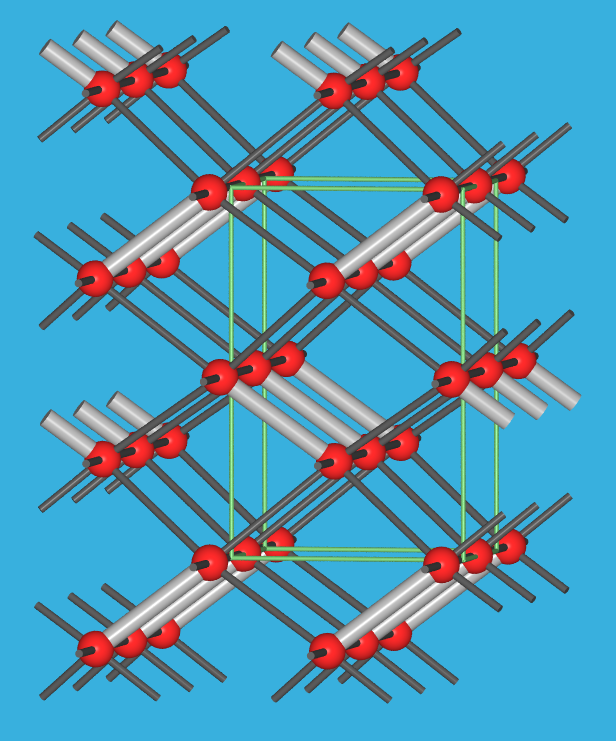
|
| Abb. 2.2.3.11. Die monokline Kristallstruktur von Te-II. Dicke, helle Bindungen sind etwas kleiner als 300 pm, die dünneren Stäbe entsprechen Te-Te-Abständen zwischen 300 und 330 pm. ‣ VRML |
| d1X-X | d2X-X | WinkelXXX | Diederwinkel | Anordnung | Bandlücke | |
| intram. [pm] | interm. [pm] | [o] | [o] | [eV] | ||
| α-S8 | 205 | 380 | 98 | cisoid | ||
| S12 | cisoid/transoid | |||||
| S∞ | transoid | |||||
| Se (grau) | 237 | 344 | 103.1 | transoid | 2.2 | |
| Te | 283 | 349 | 101.5 | transoid | 0.33 | |
| α-Po | 336 | 336 | 90 | 90 | - | 0 |
Die Abstände X-X im Molekül (intramolekular) und dazwischen (intermolekular) nähern sich nach unten im Periodensystem immer mehr an. Zugleich nimmt der metallische Charakter zu, die Bandlücke nimmt ab. Die Grenze kovalenter Bindungskonzepts ist beim Polonium erreicht. Innerhalb einer Gruppe gilt die Druckhomologenregel, nach der häufig die Hochdruckmodifikationen eines Elementes die Struktur des schwereren Homologen einnehmen.
... und weiter mit den Pnicogenen (Pentelen) (Kap. 2.2.4.) ...
| ⇦ | Inhalt | Einleitung | Kovalente FK | Metalle | Ionenkristalle | Literatur | ⇨ |
| cr_home | Metalle | Nichtmetalle | FK-Chemie | Methoden | Interm. Phasen | Oxide | Silicate | Strukturtypen |
{kind=link}
{kind=link}