- Neutrale Liganden sind z.B. H2O, NH3, PH3, ...
- Anionische Liganden sind z.B. NH2-, NH2-, N3-, CN-,....
- 1: (terminal) einfachster und häufigster Fall
- 2: einfacher Brückenligand, μ (kurz für μ2; s. (a) bis (c) in Abb. 8.3.1.
- 3: μ3-Liganden verknüpfen drei Metallatome/ionen, s. (d) und (e) in Abb. 8.3.1.
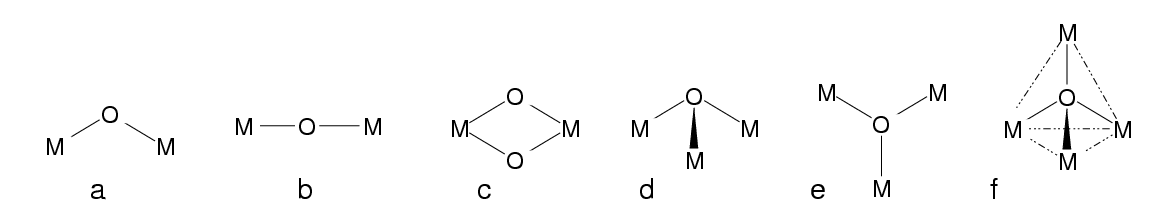
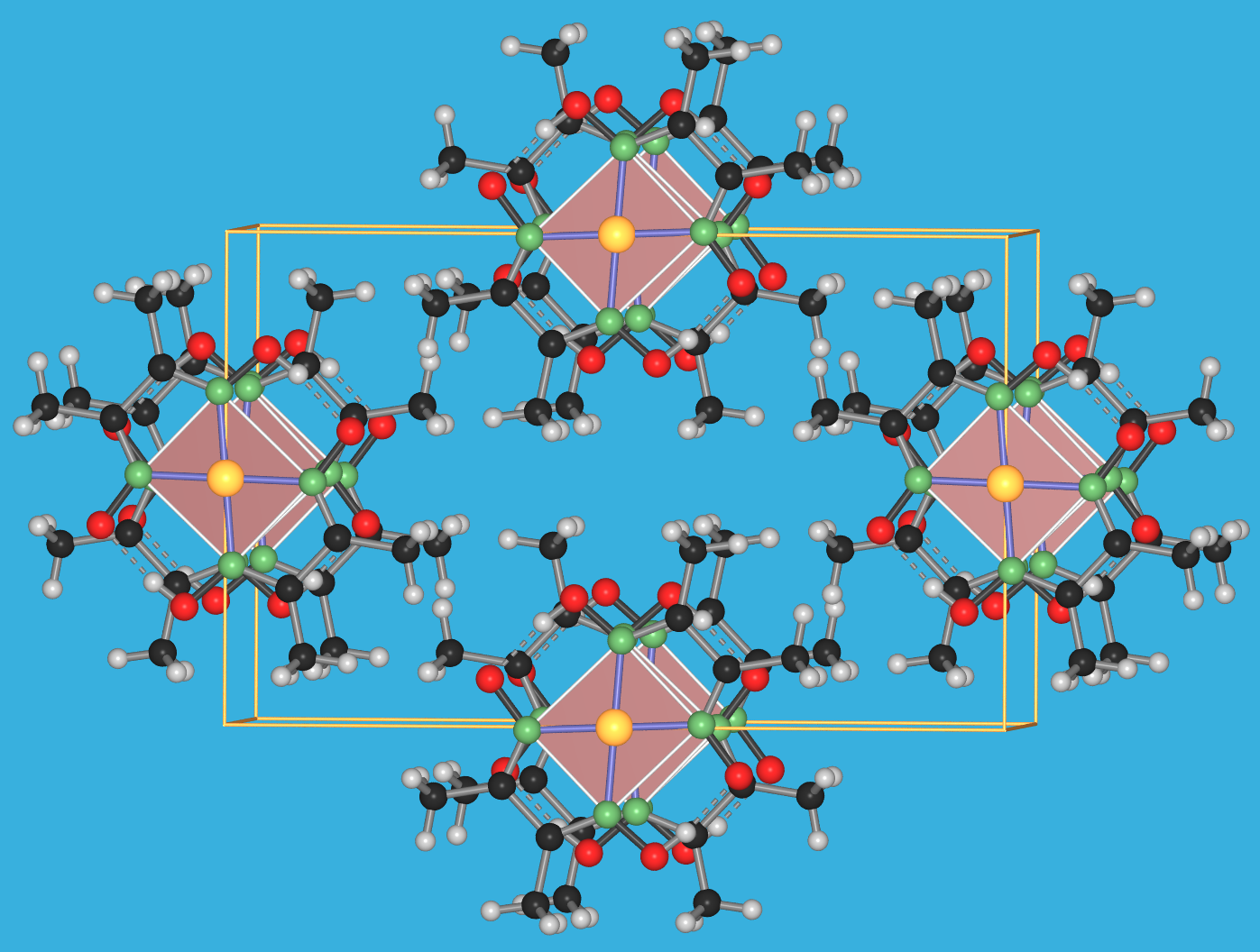
Abb. 8.3.1. Sauerstoff als Brückenligand ‣SVG
Beispiele:- a) [O3CrOCrO3]2-
- b) [Cl5RuORuCl5]4-
- c) [(H2O)2OMoO2MoO(H2O)]2+
- f) OBe4(NO3)6
- Die Ligandenkoordination ist besonders groß, wenn L als zentrales Nichtmetallatom in Clustern fungiert (f).
- n-Komplexe: Die Bindung zum M- Atom/Ion wird von nichtbindende Elektronenpaaren gebildet (häufigster Fall).
- π-Komplexe: Besetzte π-Orbitale sind für die koordinative Bindung verantwortlich.
Wichtige Liganden dieses Typs sind z.B. Ethen (1 Elekronenpaar),
Butadien (2 Elektronenpaare), Cyclopentadienyl (cp-, 3 Elektronenpaare),
Benzol (ebenfalls 3 Elektronenpaare) oder Cyclooctatetraen (cot, 2 bis maximal 4 Elektronenpaare).
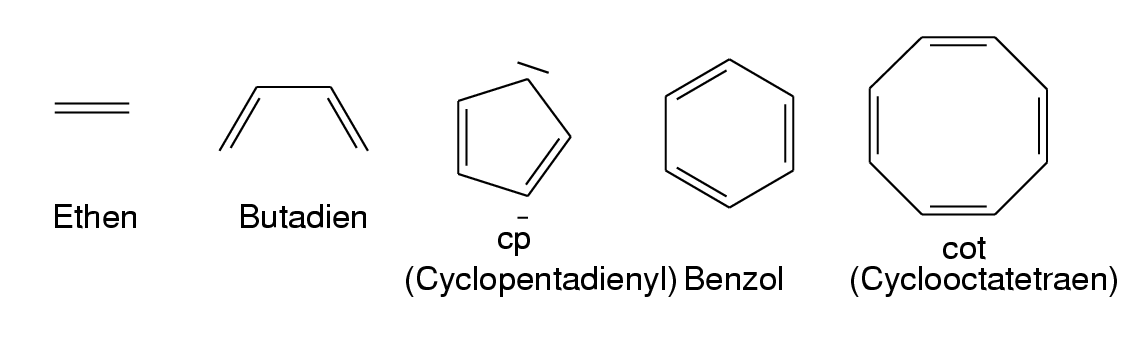
Abb. 8.3.2. Beispiele für π-Liganden ‣SVG - σ-Komplexe: Hier binden normale σ-Elektronenpaare. Wichtigstes Beispiel sind H2 (Side-on)-Komplexe.
- Für einzähnige Liganden (η1), bei denen nur zwei Elektronen, d.h. ein Elektronenpaar, bindet, gibt es tausende von Beispielen (z.B. NH3, SCN- ...).
- Einfache mehrzähnige Liganden,
bei denen mehrere Elektronenpaare zur Bindung beitragen,
können nach der Zähnigkeit weiter eingeteilt werden:
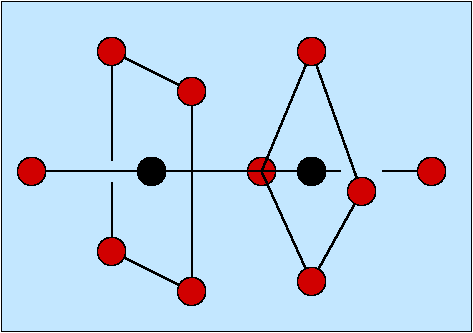
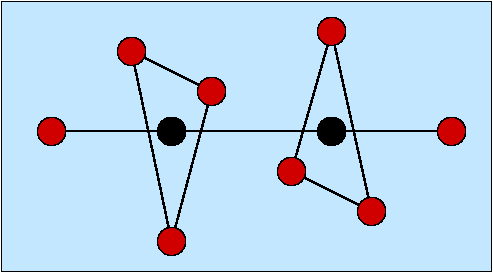
- Zweizähnige Liganden (η2; Beispiele s. Abb. 8.3.3.) weisen zwei Donorzentren auf.
Günstig ist es dabei,
wenn der Abstand zwischen diesen Atomen vier oder fünf Atome beträgt, weil
dann bei der Komplexbildung Fünf- bzw. Sechs-Ringe entstehen. Einer
der einfachsten Liganden ist Ethylendiamin (en).

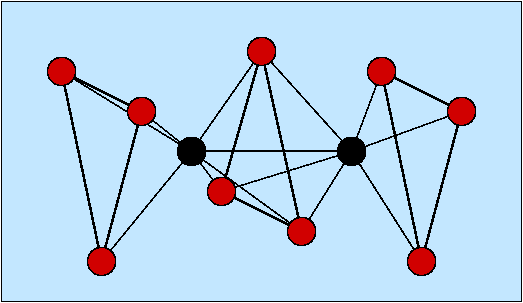
Abb. 8.3.3. Beispiele für zwei-zähnige Liganden ‣SVG - Dreizähnige Liganden (Beispiele s. Abb. 8.3.4.) weisen entsprechend
drei Donorzentren auf. Einfache Beispiele sind 'Terpyridin' (ter) oder
Diethylentriamin (dien).
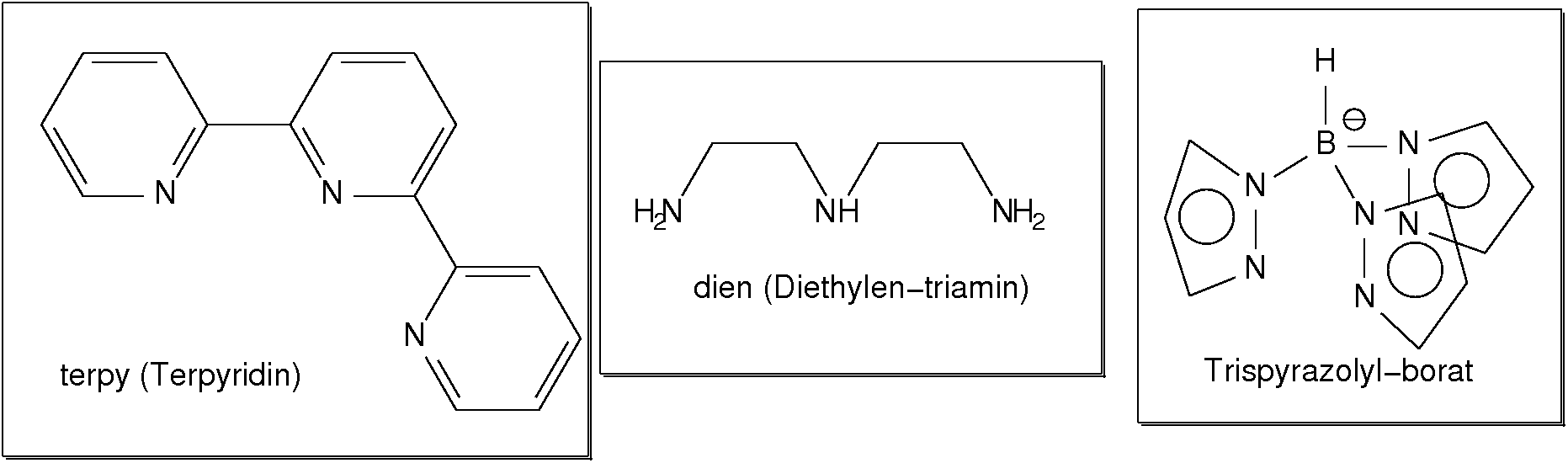
Abb. 8.3.4. Beispiele für einfache dreizähnige Liganden ‣SVG - Einfache vierzähnige Liganden sind wieder die Ketten-Amine (hier 'trien').
Ein interessanter, ebenfalls vierzähliger Ligand mit bereits vorgeprägter quadratisch-
planarer Konfiguration ist 'salen' (Bis(salicyliden)ethylendiamin).
In der Komplexometrie (s.u.) wird der vierzähnige sog. tripodale Ligand
Nitrilo-triessisäure (NTA, Titriplex I) eingesetzt.
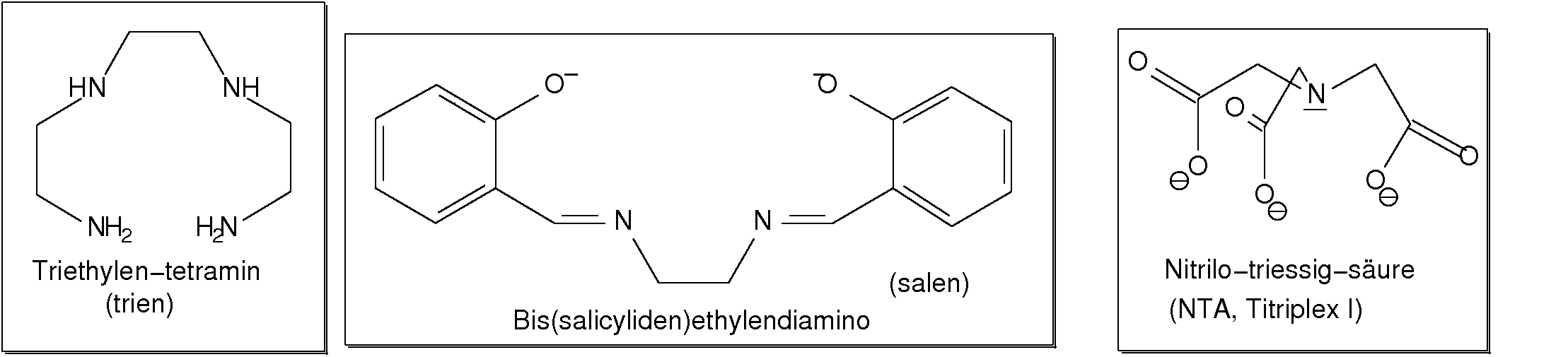
Abb. 8.3.5. Beispiele vierzähniger Liganden ‣SVG - Der sicher bekannteste einfache sechszähnige Ligand ist Ethylendiamin-tetraacetat (EDTA),
der ebenfalls als Komplexon in der Analytik verwendet wird (freie Säure: Titriplex II;
Dihydrat des Dinatriumsalzes: Titriplex III).
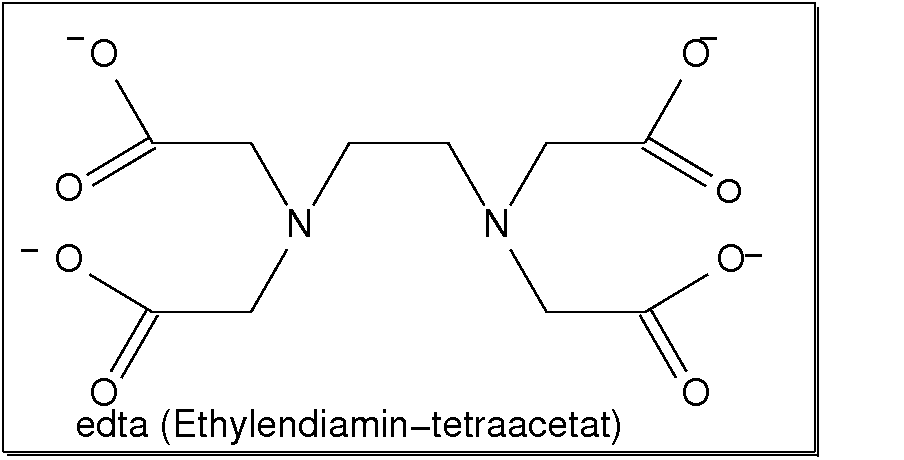
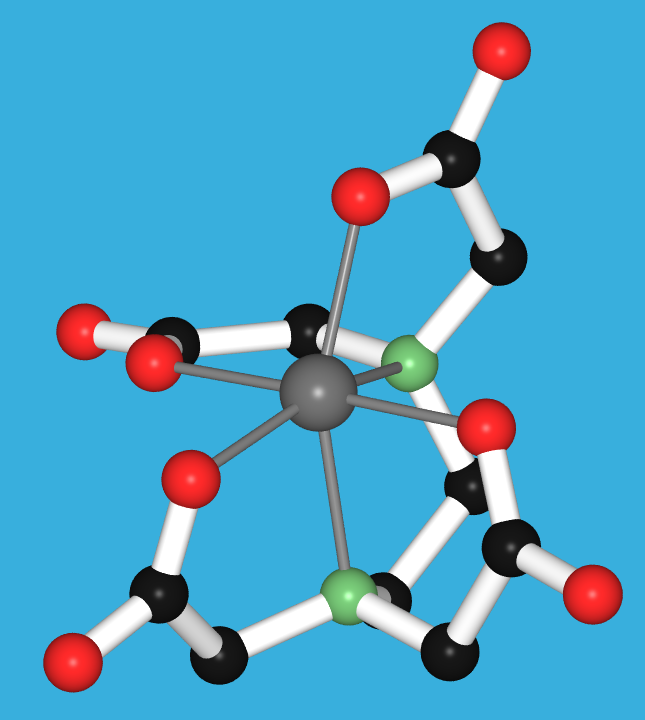
Abb. 8.3.6. EDTA: ein Beispiel für einen sechszähnigen Liganden ‣SVG bzw. ‣VRML
⚗ Chelateffekt: Auflösen von Kalk mit EDTA (20MB|MP4|H264) Gefälltes Calciumcarbonat (CaCO3, Kalk!) löst sich in EDTA, da die Ca2+-Ionen den sehr stabilen Chelatkomplex mit diesem sechszähnigen Liganden (s. Abb. 8.3.6. oben) bilden. - Zweizähnige Liganden (η2; Beispiele s. Abb. 8.3.3.) weisen zwei Donorzentren auf.
Günstig ist es dabei,
wenn der Abstand zwischen diesen Atomen vier oder fünf Atome beträgt, weil
dann bei der Komplexbildung Fünf- bzw. Sechs-Ringe entstehen. Einer
der einfachsten Liganden ist Ethylendiamin (en).
- Neben diesen einfachen mehrzähnigen Liganden gibt es eine Reihe weiterer spezieller
Chelatliganden:
- Ringförmige Liganden bilden bei der Komplexbildung gleich mehrere günstige
Ringe zum Metall (Makrozyklischer Effekt). Besonders bekannt sind hier
die 'Kronenether', die durch Variation der Ringgrössen z.B. selektiv für die
Komplexierung unterschiedlich gro�er Alkalimetallkationen eingesetzt werden können.
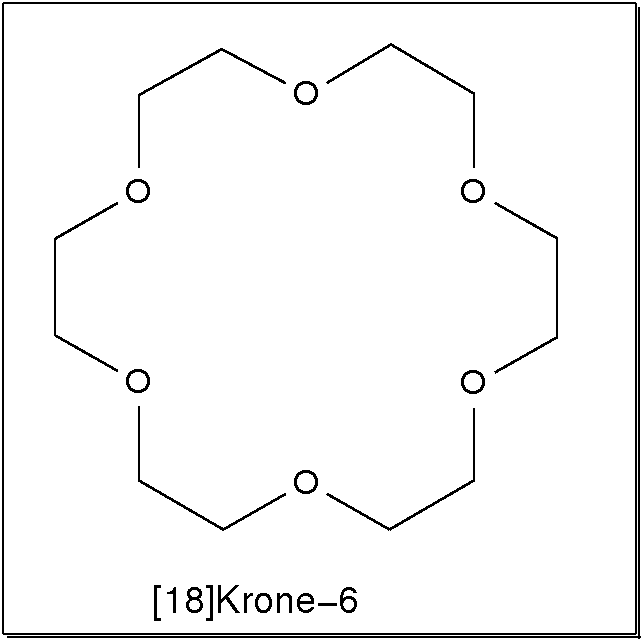
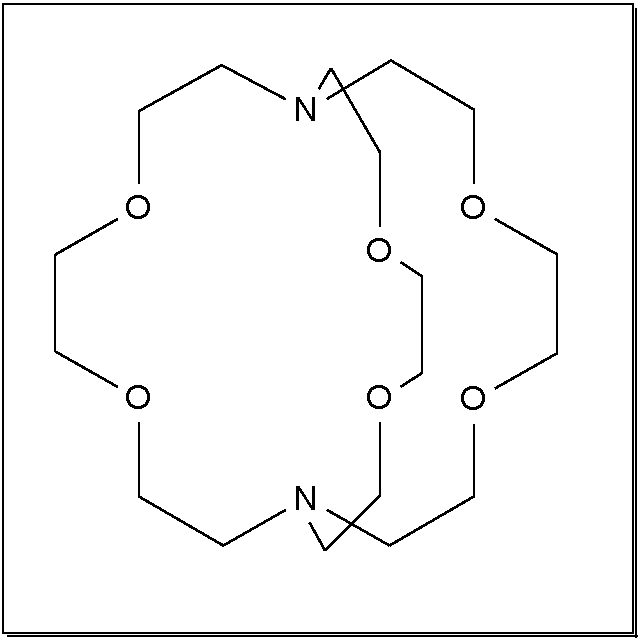

Abb. 8.3.8. [18]Krone-6 als Beispiel für makrozyklische Polyether ‣SVG Abb. 8.3.9. 2.2.2.crypt, ein Kryptand ‣SVG bzw. ‣VRML - Liganden mit starrer Grundgeometrie prägen dem Komplex die Geometrie auf. Sie
müssen zur Komplexbildung nicht umgebildet werden und sind für Anwendungen, Biologie,
Medizin, ... sehr wichtig. Eine große Gruppe sind hier die Tetrapyrrol-Liganden wie
z.B. Porphyrin.


Abb. 8.3.10. Porphyrin: Grundkörper der zahlreichen Tetrapyrrol-Liganden (‣SVG) (links) und Foto des neutralen Kupfer(II)-Komplexes, der als 'Brilliant-Blau K' ein sehr blaues Blaupigment ist (z.B. Kugelschreiber-Farben, rechts).
- Ringförmige Liganden bilden bei der Komplexbildung gleich mehrere günstige
Ringe zum Metall (Makrozyklischer Effekt). Besonders bekannt sind hier
die 'Kronenether', die durch Variation der Ringgrössen z.B. selektiv für die
Komplexierung unterschiedlich gro�er Alkalimetallkationen eingesetzt werden können.
Komplexe mit solchen mehrzähnigen Liganden haben aufgrund des sogenannten Chelateffekts eine höhere Stabilität. Einmal handelt es sich um einen kinetischen (Reaktionsmechanismus) und um einen thermodynamischen Beitrag (Entropie-Effekt, d.h. erhöhte Entropie) gemäß dem chemischen Gleichgewicht:
Besonders die Bildung von 5-fachen Chelatringen ist besonders günstig. Bekanntes Beispiel für einen Komplex mit sechszähnigem Liganden sind die aus der Analytik bekannten edta-Komplexe (s. Abb. 8.3.6.) In Ticp2S5 liegt auch mal ein Sechsring vor.

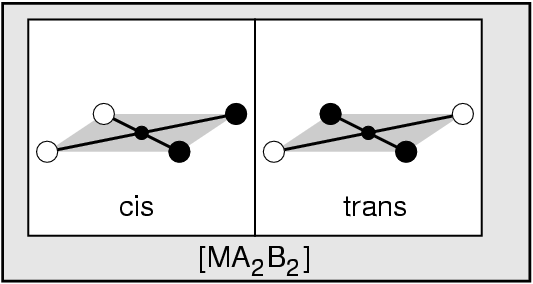
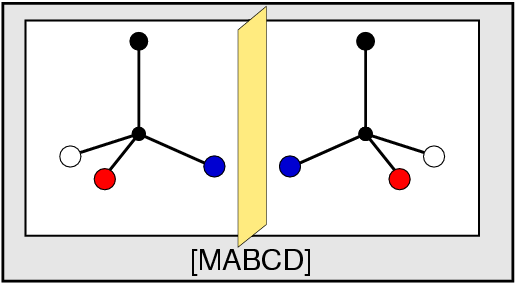
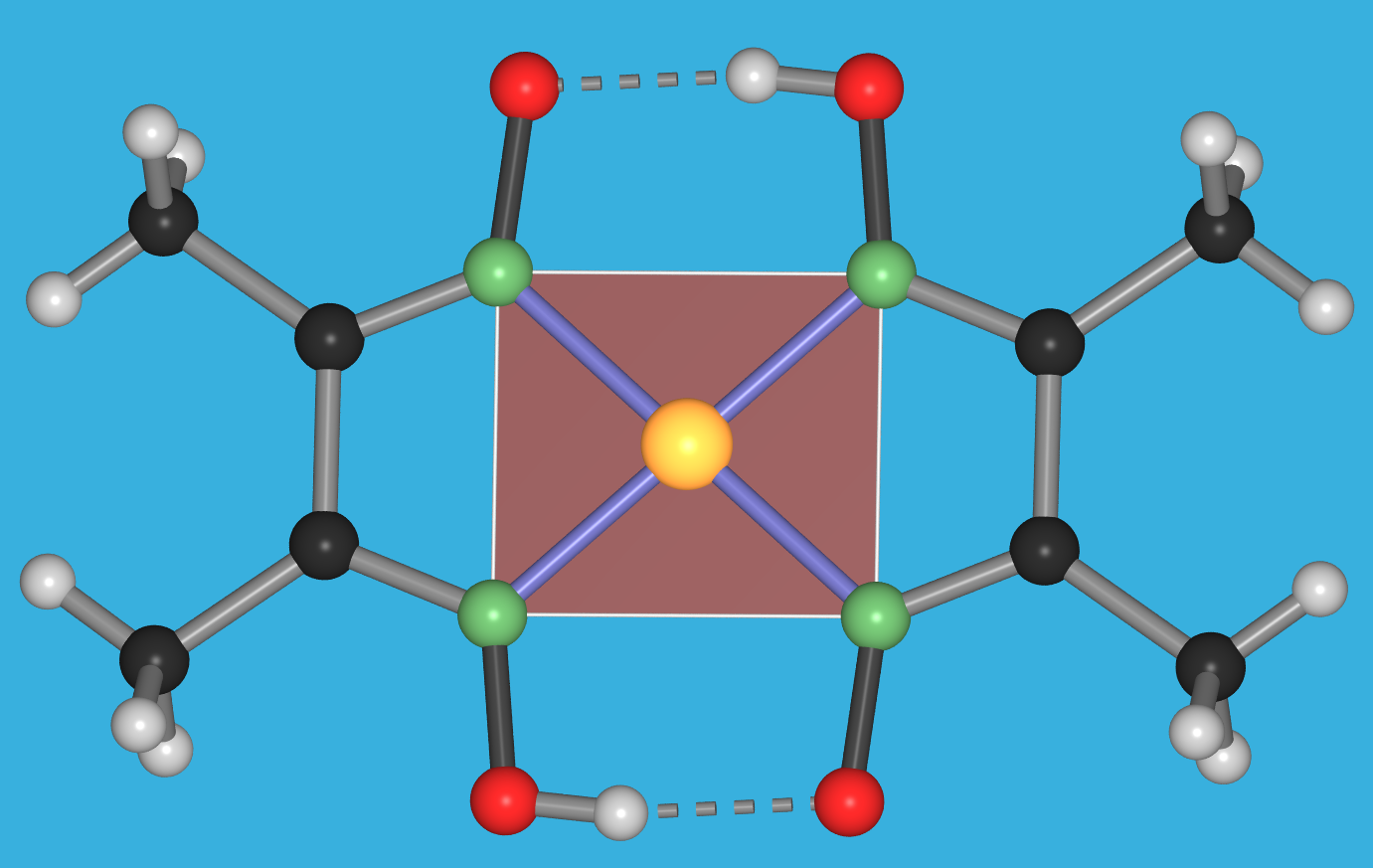
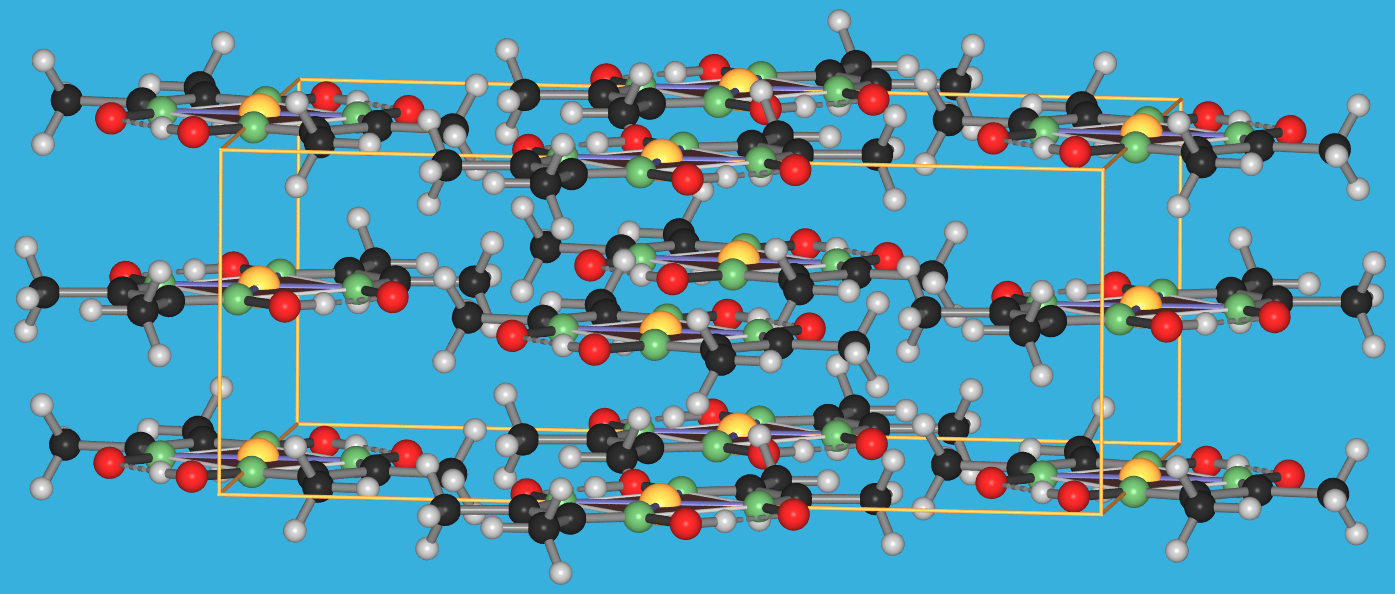


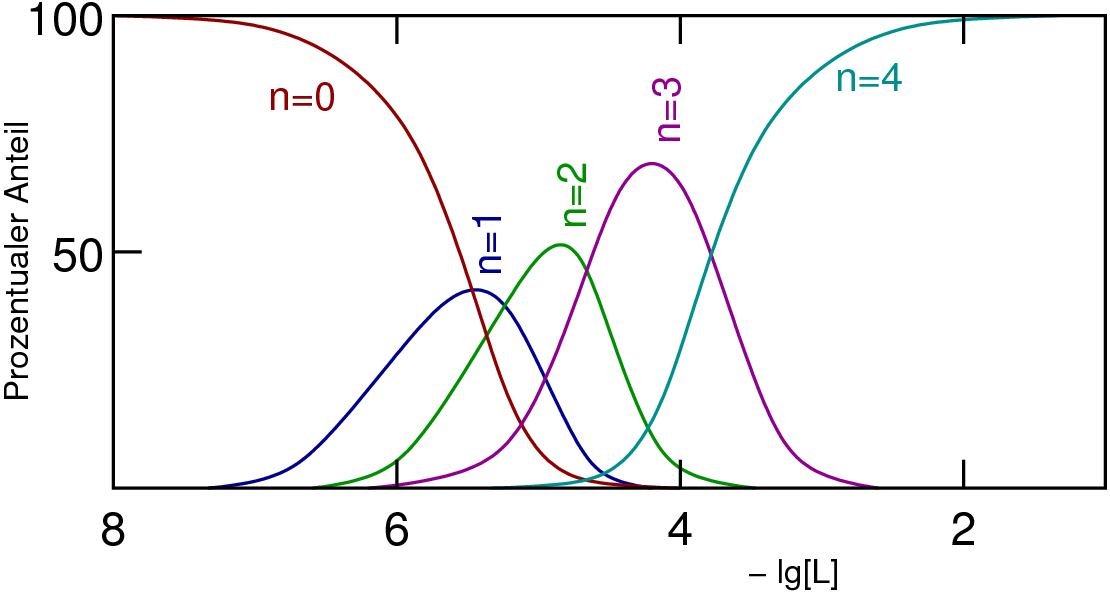
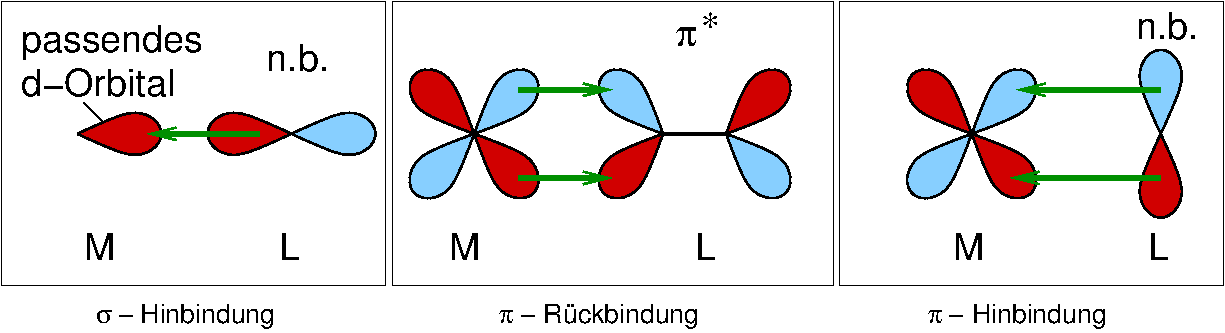
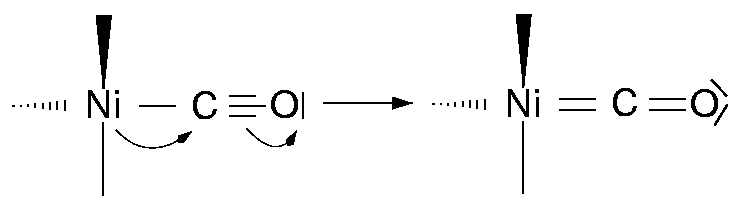
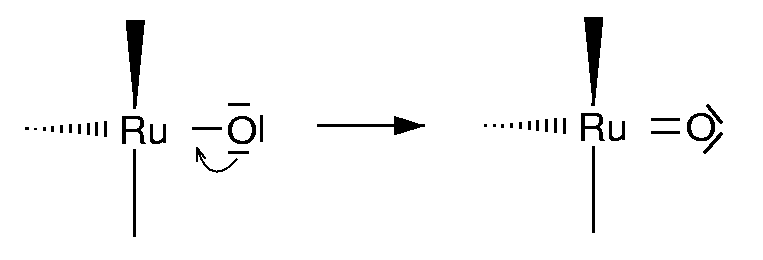
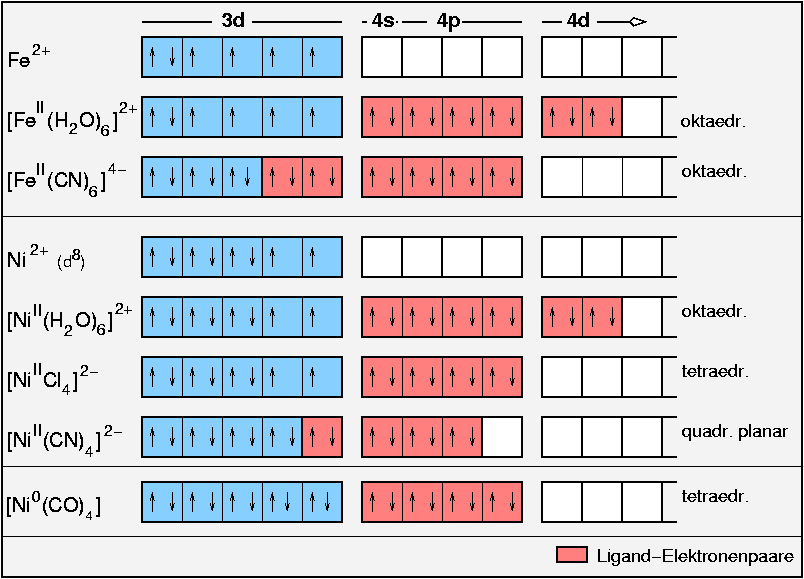
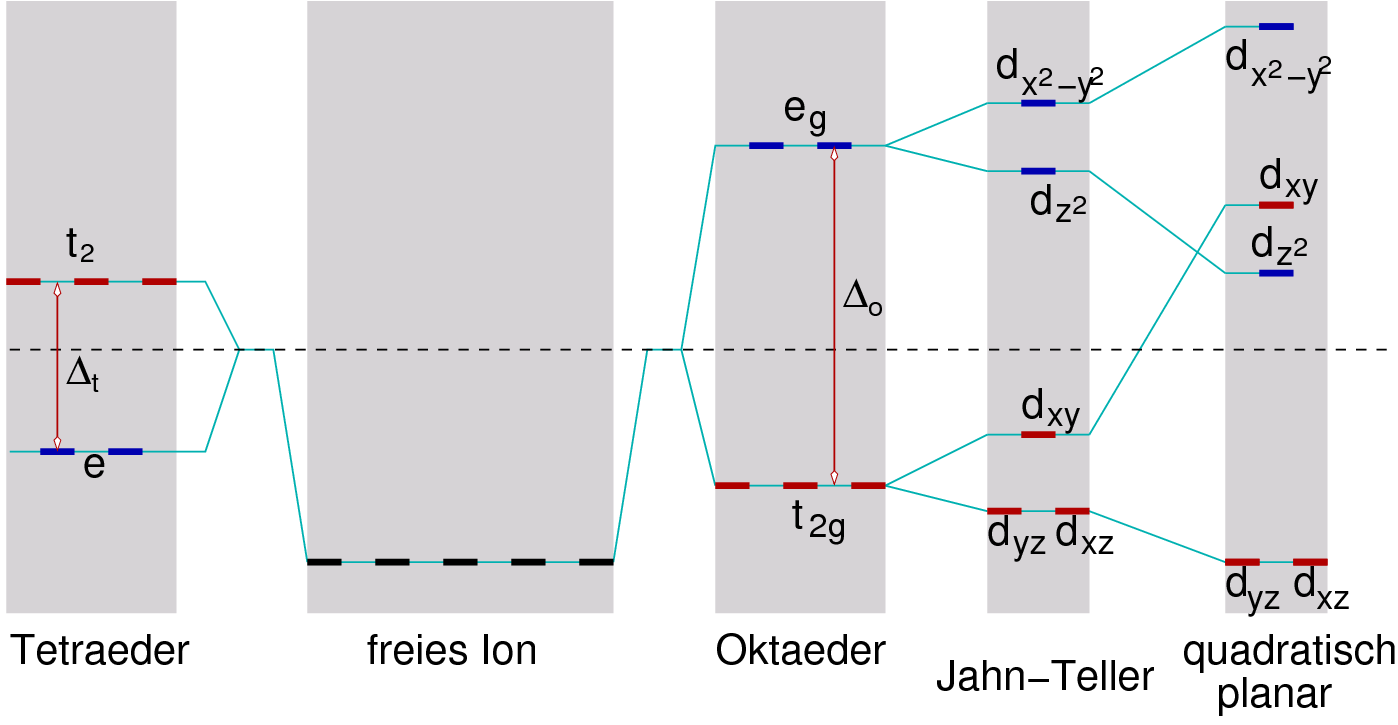
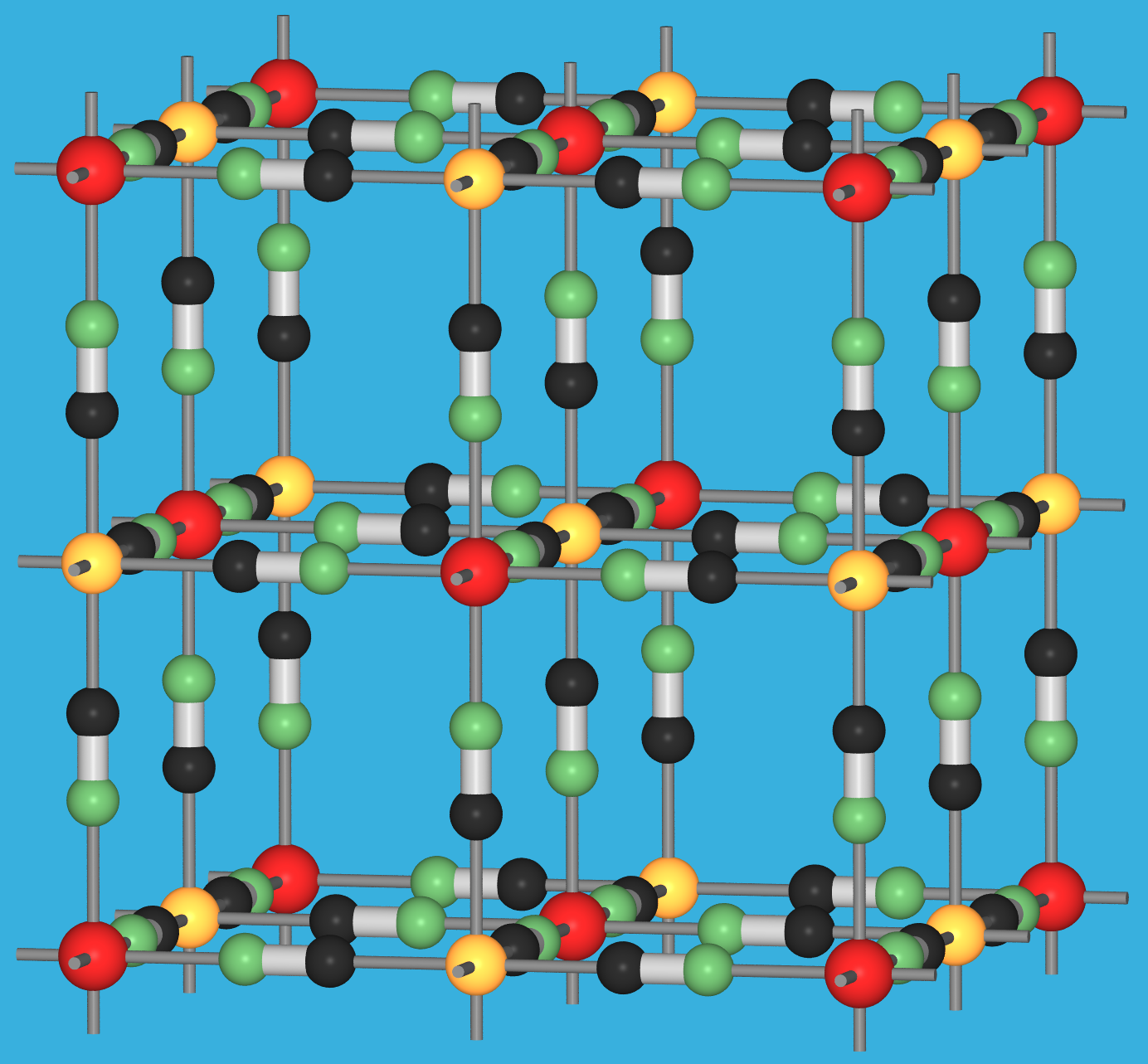
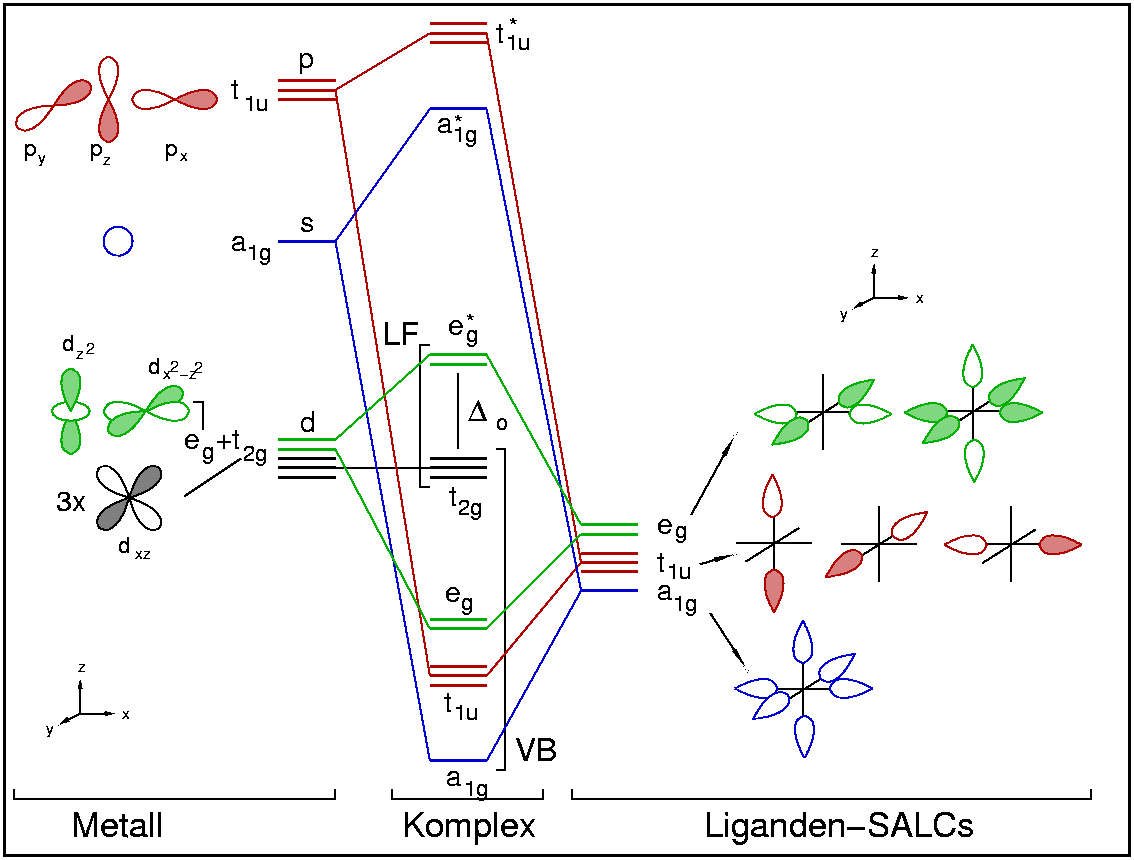

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}