Vorlesung Strukturchemie der Oxide
3. Binäre Metalloxide
3.3. Normale Metalloxide
Bevor die wichtigsten Strukturtypen der Metalloxide
im Detail vorgestellt werden, soll
zunächt ein allgemeiner Überblick über
physikalische Eigenschaften, die chemische Bindung und die wichtigsten
strukturbestimmenden Faktoren gegeben werden.
Die meisten Metalloxide etwa bis zur Oxidationsstufe +4
bilden echte Ionenkristalle (und damit demnach dreidimensional zu beschreibende
Verbände).
Bei den Übergangsmetalloxiden höherer Oxidationsstufen
finden sich dann eher kovalent aufgebaute Verbindungen mit
niederdimensionalen Verbänden.
Beispiele hierfür sind molekulare Verbindungen wie Mn2O7
oder RuO4, Kettenstrukturen wie CrO3
und Schichten in V2O5, MoO3 oder
Re2O7.
Nicht nur strukturell sondern auch aufgrund der physikalischen
Eigenschaften sind die Metalloxide damit bei weitem keine einheitliche
Klasse. Beispielweise variieren die Schmelzpunkte
von sehr hochschmelzenden Verbindungen wie z.B. HfO2 (Mp 2800
oC) und den molekularen Phasen wie z.B.
Mn2O7 (Mp. 6oC) stark.
Auch die elektrischen Eigenschaften reichen von Isolatoren (MgO)
bis zu guter metallischer Leitfähigkeit (ReO3).
Viele Übergangsmetalloxide zeigen darüberhinaus interessante physikalische Eigenschaften:
Z.B. sind EuO und CrO2 ferromagnetisch,
viele Übergangsmetall-Oxide MO zeigen anormales elektrisches und
magnetisches Verhalten (z.B. NiO).
Aufgrund dieser Eigenschaften finden viele Oxide ausgedehnte technische Verwendung
(z.B. CrO2 für Magnetbänder).
Aber auch als Grundstoffe in der Großindustrie sind Metalloxide von sehr großer
Bedeutung. Beispiele hierfür sind die Verwendung als Pigmente (z.B.
TiO2), Füllstoffe oder Absorber-Materialien (z.B.
Al2O3).
Strukturell lassen sich alle Metalloxide, die
nicht durch gerichtete Bindungen bestimmt werden (das ist der Hauptteil!)
wie viele andere Ionenkristalle durch die Füllung von oktaedrischen
und tetraedrischen Lücken in dichtesten Packungen beschreiben,
da der Ionenradius von O2- mit 140 pm deutlich grösser ist
als die Radien der meisten Metallkationen.
Abweichungen von diesem Konzept treten bei den Übergangsmetallen auf,
wo zusätzlich zu rein geometrischen Verhältnissen Kristallfelder
eine Rolle spielen.
Besonders bei den Übergangsmetalloxiden finden sich
neben den Daltoniden (stöchiometrische Verbindungen) oft nichtstöchiometrische
Phasen (z.B. W-Oxide, Fe-Oxide.).
Die untenstehende Tabelle gibt eine Übersicht über alle
stöchiometrischen Metalloxide.
Geordnet nach Oxidationsstufe sind die jeweils auftretenden Strukturtypen
angegeben. Auf die Angabe von gemischtvalenten Phasen und Phasenbreiten
muß hierbei verzichtet werden.
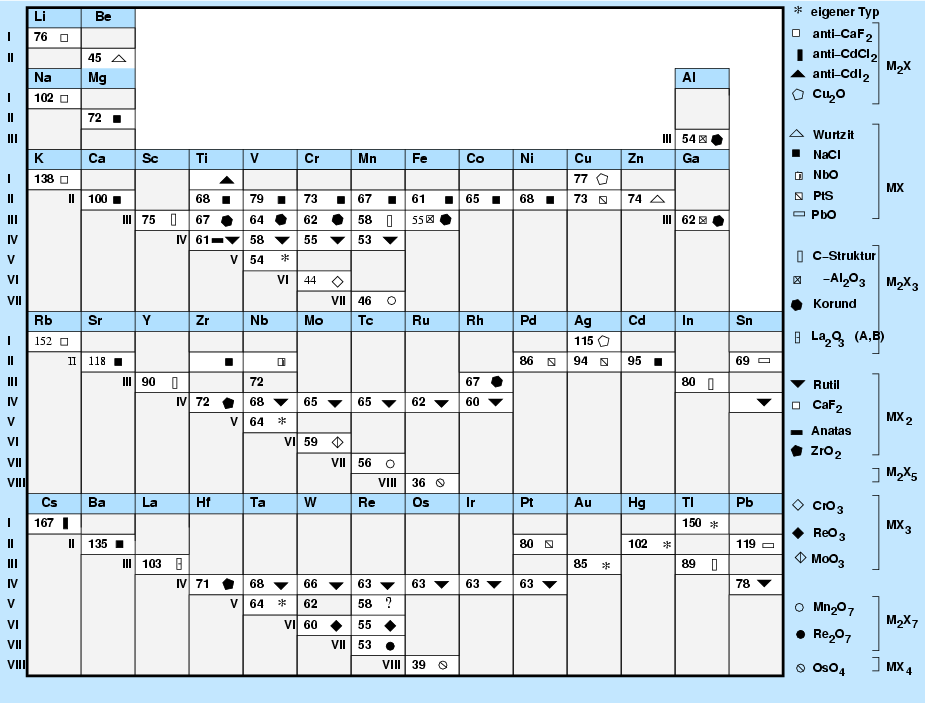
|
| Abb. 3.3.1. Struktur der Metall-Oxide im Periodensystem |
Die Symbole in Abb. 3.3.1. bedeuten im Einzelnen
- Quadrate oder Rechtecke bezeichnen Strukturen auf f.c.c.-Basis von O2--Anionen.
- Dreiecke oder Sechsecke bezeichnen Strukturen auf h.c.p.-Basis von O2--Anionen.
- Offene Symbole stehen für gefüllte Tetraederlücken.
- Geschlossene Symbole bezeichnen gefüllte Oktaederlücken.
- Runde Symbole oder Rauten kennzeichnen Verbindungen mit deutlichem kovalentem Bindungsanteil.
Vor der Besprechung der Oxiden im Einzelnen
sollen zunächst die strukturbestimmenden Faktoren und die chemische Bindung
in Ionenkristallen kurz wiederholt werden ( 3.3.1)
Die systematische Strukturklassifizierung auf der Basis dichter Packungen
folgt unter 3.3.2. Im Abschnitt
3.3.3 werden die wichtigsten Oxide nach Stöchiometrie im
Detail besprochen.
Die überwiegende Zahl binärer Metalloxide (bis etwa zur Oxidationsstufe
+4) sind mehr oder weniger ionisch aufgebaute Verbindungen.
Der Ionencharakter läßt sich z.B. nach Pauling
(gemäß 1 - e-1/4(xA-xB))
ermitteln und liefert die folgenden Werte:
| Verbindung |
|
LiF |
MgO |
FeO |
OsO4 |
| Ionencharakter nach Pauling |
|
0.9 |
0.73 |
0.52 |
0.35 |
Qualitative Betrachtung: Ionische Verbindungen
werden durch elektrostatische Kräfte zusammengehalten;
die Anionen sind von Kationen umgeben und umgekehrt.
Eine große elektrostatische Anziehung, d.h. eine große Gitterenergie
wird dabei durch hohe Koordinationszahlen erreicht.
Kontakte der Anionen bzw. Kationen untereinander sollten minimal, Kontakte
zwischen Anionen und Kationen maximal sein.
Gleichzeitg muß lokale elektrostatische Neutralität erreicht sein.
Quantitative Betrachtung
Bei kleinen Drücken verbleibt von der
Gibbs-schen freien Energie (G = E - TS + PV)
nur die Helmholtz-Energie als wesentlicher Beitrag: F = E - TS.
Die innere Energie E des Kristalls besteht dabei aus zwei Anteilen
(E = UL + EV),
dem dynamischen Anteil EV und der
statischen Gitterenergie UL.
Diese Gitterenergie ist wiederum die Summe verschiedener Anteile:
(in Klammern typische Prozentangaben zum Beitrag an der gesamten
Gitterenergie:
UL = Ea (80%) + Er (10-15%)
+ Es (5-10%) + kovalente Bindungsanteile + van der Waals
+ ..
Die Anteile sind (Z=Ladungen, R=Gleichgewichtsabstand, B=Proportionalitätskonstante)
- Ea: elektrostatische Anziehung
(Ea = - (Z1 Z2 e2) / R )
- Er: elektrostatische Abstoßung
(Er = B / Rn mit n = 5-12)
- Es Kristallfeldstabilisierungsenergie (nur bei den Übergangsmetallen)
Ea und Er zusammen hängen
(unter Vernachlässigung der Ligandenfeldstabilisierungsenergie)
nur vom Abstand der Ionen, deren Ladungen sowie dem Strukturtyp ab:
Ea + Er = UL = - [ { A N0
z1 z2 e2} / {Re} ] (1- 1/n)
Typische Werte für die Madelungkonstante A sind:
| Strukturtyp |
|
NaCl |
Rutil |
Anatas |
Fluorit |
| Madelungkonstante A |
|
1.76 |
2.408 |
2.400 |
2.519 |
UL kann experimentell z.B.
auf der Basis des Born-Haber-Kreisprozeßes bestimmt werden.
Danach ist die treibende Kraft für die Bildung eines Ionenkristalls
(d.h. die Bildungsenthalpie) zusammengesetzt aus:
ΔHf = ΔHM + ΔHx + ΔHIE + ΔHEA + UL
wobei:
- Hf: Bildungsenthalpie
- HM: Sublimationsenergie
- Hx: Dissoziationsenergie
- HIE: Ionisierungsenergie
- HEA: Elektronenaffinität
- Gitterenergie: UL
Einige Werte für die Gitterenergien:
| Verbindung |
|
NaCl |
MgO |
BaO |
| Gitterenergie UL |
|
-760 kJ/mol |
-4000 kJ/mol |
-3200 kJ/mol |
| Schmelzpunkt [oC] |
|
800 |
2800 |
1900 |
Man sieht, daß die Schmelzpunkte etwa parallel mit
der Gitterenergie variieren.
Die qualitative Folgerung für die maximale Gitterenergie ist, daß
die Kationen von möglichst vielen Anionen umgeben sein sollten (und
umgekehrt) und Kationen und Anionen �zueinander passen� sollen.
In der Strukturchemie von Ionenkristallen gibt es hierzu einige
empirische Regeln (sog. Pauling-Regeln):
1. Pauling'sche Regel
Für die Polyeder um die Kationen gilt, daß
der Abstand Kation-Anion durch die Summe,
die Koordinationszahlen durch das Verhältnis der Ionenradien bestimmt wird
(sog. Radienverhältnisregel).
Demnach bestimmt also das Radienverhältnis wesentlich die Koordinationszahl
und damit auch den möglichen Strukturtyp.
Konkret folgt für die Koordinationszahlen der Kationen in Oxiden
(rein geometrische Betrachtung, Ligandenfelder vernachlässigt, Radius von
O2-
= 140 pm):
| |
|
CN=4 (Tetraeder) |
CN=6 (Oktaeder) |
CN=8 (Würfel) |
| Radienverhältnis (untere Grenze) |
|
0.225 |
0.414 |
0.732 |
| Kationenradius in Oxiden [pm] |
|
36 |
58 |
102 |
| Zusammensetzung |
|
Strukturtypen |
|
|
| A2B |
|
anti-CaF2 |
- |
- |
| AB |
|
ZnS (Zinkblende, Wurtzit) |
NaCl |
CsCl |
| AB2 |
|
SiO2 |
Rutil, CdCl2, CdI2 |
CaF2 |
Die Strukturen lassen sich zum großen Teil
wegen der Ionenradien als dichte Packungen aus O2--Ionen
mit gefüllten Lücken beschreiben.
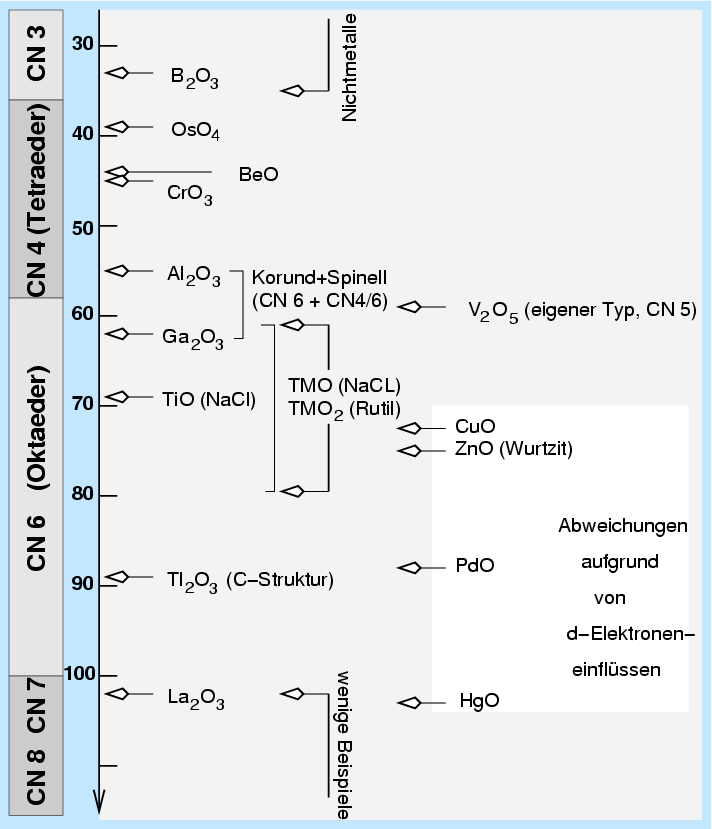
|
| Abb. 3.3.2. Radienverh"altnisregel an Beispielen |
Die Überprüfung der Radienverhältnisregel an Beispielen (s. Abb. 3.3.2.)
zeigt, daß sie nur bedingt gilt.
Abweichungen
finden sich vor allem bei den späten Übergangsmetallen (Cu,Zn)
und den Alkali- und Erdalkalimetallen.
Ein Teil dieser Abweichungen wird verständlich, in den Fällen,
in denen die innere Elektronendichte eines Ions nicht kugelsymmetrisch ist.
Hier sind zwei Fällen denkbar:
- Inert-Pair Einfluß für Hauptgruppenmetalloxide mit (maximaler Oxidationszahl-2)
- Ligandenfeldeinfluß für nicht vollbesetzte d-Schalen (bei den Übergangsmetallen)
zu 1: Inert-Pair Einfluß
Bei Hauptgruppenmetalloxiden mit der (maximalen Oxidationsstufe-2)
(z.B. Sn2+, Pb2+)
kommt es zu einer Verzerrung der Metall-Sauerstoff-Koordination durch das
einsame Elektronenpaar.
Z.B. sind im Pb2+-Oxid die 6 s2-Elektronen
stereochemisch aktiv (sp-Hybrid), so daß PbO und und auch SnO eine tetragonal verzerrte
CsCl-Struktur ausbilden.
zu 2: Ligandenfeldeinfluß
In die Gitterenergie UL gehen bei Übergangsmetallionen mit
unvollständig gefüllten d-Elektronenschalen neben den elektrostatischen
Anteilen Ea und Er (anziehender und abstoßender
Teil) und kovalente Bindungsanteile, van der Waals Anteilen usw. auch
Ligandenfeldstabilisierungsenergien (LFSE, Es) ein. Die Gitterenergie ist damit in
diesen Fällen entsprechend größer.
Der Einfluß der d-Elektronen auf das Koordinationspolyeder um
Übergangsmetall-Kationen ist aus der Komplexchemie bekannt:
aus der Ligandenfeld-Theorie folgt, daß die Ligandenfeldstabilisierungsenergie (LFSE)
bestimmt, welches Ion welche Koordination (hier gegen den Liganden
O2-) bevorzugt.
Während sich in den isolierten Kationen alle d-Orbitale auf dem gleichen
energetischen Niveau befinden, wird
bei der LF-Theorie
der Einfluß
der Liganden (die zunächst als Punktladungen betrachtet werden)
auf die Energieniveaus der verschiedenen d-Orbitale
betrachtet. Beispielsweise geben sich im oktaedrischen und im tetraedrischen
Ligandenfeld Aufspaltungen der d-Orbitale in zwei Gruppen:
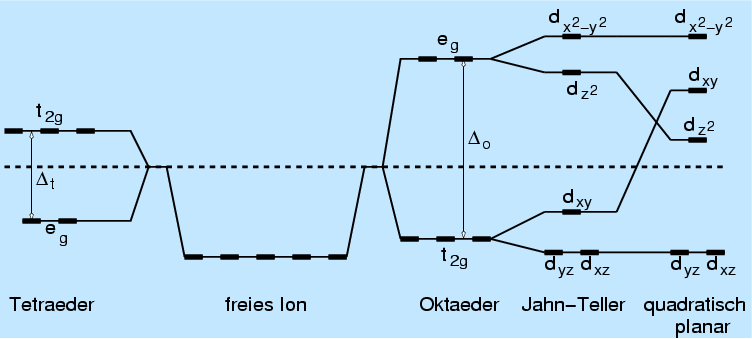
|
| Abb. 3.3.3. Ligandenfeldabspaltung in nichtkugelsymmetrischen Kristallfeldern |
- Die dxy-, dxz- und dyz-Orbitale weisen auf die
Diagonalen des Koordinatensystems (t2g-Zustände).
- Das dx2-y2- und das
dz2 liegen dagegen auf den Koordinatenachsen
(eg-Zustände).
Im oktaedrischen Feld werden entsprechend der Abstoßung Ligand - d-Zustand
die t2g Zustände entsprechend dem Schwerpunktsatz energetisch erniedrigt,
während im tetraedrischen Ligandenfeld die Verhältnisse genau umgekehrt
sind. Die Größe der Energieaufspaltung wird sowohl von der Art des
Liganden als auch vom Metallatom bestimmt.
Generell gilt, daß die Aufspaltung im oktaedrischen Ligandenfeld
etwa doppelt so groß ist wie die im tetraedrischen Fall.
Die Aufspaltung im oktaedrischen Feld
(Δo) liegt allgemein im Bereich von 70 bis 350 kJ/mol,
bei Oxiden bei 70-160 kJ/mol. Sie wird bestimmt durch:
- die Ladung von M (groß bei hoher Ladung)
- die Art der d-Orbital (3d < 4d < 5d)
- den Abstand Metall-Ligand (proportional 1/R)
- die Art des Liganden.
Die sog. spektrochemische Reihe: (I- < S2- <
F- = O2- < ... < OH-) zeigt, daß
O2- ein eher schwacher Ligand ist, so daß Oxide im allgemeinen High-Spin-Kationen
enthalten).
Die Kristallfeldstabilisierungsenergie (die in die Gitterenergie direkt mit
eingeht) ist die Energie, die durch Einfüllen der Elektronen in die
jeweils niedrigeren Niveaus gewonnen wird.
Die Besetzung der Zustände folgt i. A. dem Pauli-Prinzip,
die Spin-Paarungsenergie bestimmen, ob High-Spin oder Low-Spin-Systeme
gebildet werden. Da die Aufspaltung im oktaedrischen Feld etwa doppelt
so groß ist, wie die im tetraedrischen Feld, wird bei Kationen
mit d3, d4, d6, d7 und
d8-Elektronenkonfiguration i.A. die oktaedrische Umgebung
bevorzugt (sog. Octahedral Site Preference Energy). Die folgende
Tabelle gibt einen Überblick über das Ausmaß der Bevorzugung:
| Elektronen- |
|
Oktaeder- |
Tetraeder- |
|
| konfiguration |
Ion |
Stabilisierung |
Stabilisierung |
Differenz |
| |
|
|
|
|
| d3 |
Cr3+ |
225 |
67 |
158 |
| d5 |
Fe3+ |
0 |
0 |
0 |
| d6 |
Fe2+ |
50 |
33 |
17 |
| d8 |
Ni2+ |
122 |
36 |
86 |
| d10 |
Zn2+ |
0 |
0 |
0 |
Dieser Einfluß der d-Elektronenverteilung macht sich bei den verschiedenen
Oxiden unmittelbar bemerkbar:
- Bei den Oxiden der Zusammensetzung M2O3
bildet Cr3+ nur die Korund-Struktur aus (Besetzung von
Oktaederlücken), während man bei Fe3+ außer der Korund- auch
die gamma-Al2O3-Struktur (Defekt-Spinell = Besetzung von oktaedrischen und
tetraedrischen Lücken) kennt.
- Die Oxide MO kristallisieren fast alle im NaCl-Typ (Besetzung
von Oktaederlücken). Ausnahmen sind CuO (stark verzerrtes Oktaeder) und
ZnO (Besetzung von Tetraederlücken).
Der Einfluß der Art der d-Elektronen (3d < 4d < 5d) auf Δo
zeigt sich z.B. bei den d8-Systemen: Während NiO im NaCl-Typ
kristallisiert (Oktaederkoordination), findet sich in PdO und PtO
wegen der deutlich größeren Aufspaltung quadratisch planare
Koordination.
- Die Unterschiede in den Stabilisierungsenergien sind bei
ternären Oxiden (z.B. den Spinellen) für die Verteilung der Kationen
auf den unterschiedlichen Positionen verantwortlich.
In Spinellen besetzen z.B. meist die Hauptgruppenelementionen
(Mg2+, Al3+) die tetraedrischen Lücken.
Cr3+ drängt Ni2+ auf Tetraederpositionen.
Fe3O4 bildet einen Inversspinell, d.h.
Fe2+-Ionen (d6) besetzten auch
oktaedrische, Fe3+ tetraedrische Lücken.
Daß die LFSE nicht allein für die Kationenverteilung in Spinellen
verantwortlich gemacht werden kann, zeigt das Beispiel des
Co3O4, in dem das Co3+ (d6) Ion
die Oktaeder- und Co2+ (d7) die
Tetraederposition besetzt (Normalspinell). Dieses Verhalten wird auch auf
die Neigung von Co3+ zur Ausbildung des LS-Zustands
zurückgeführt.
Zusammenfassend (Kap. 3.3.1.) läßt sich feststellen, daß
die Metalloxide i.a. als dichte Packungen von O2-
mit Kationen in den Lücken beschrieben werden können.
Die Koordinationszahl um das Kation wird vom
Radienverhältnis Anion-Kation (1.Pauling'sche Regel)
bestimmt. Die LFSE bewirkt (bei Übergangsmetallionen mit entsprechender
d-Elektronenkonfiguration) zusätzlich eine Bevorzugung der
oktaedrischen Umgebung. Weitere Details der Elektronenkonfiguration
bestimmen das Ausmaß der Jahn-Teller-Verzerrung (CuO), die im
Extremfall (PdO, PtO) bis zu quadratisch planarer Umgebung führen
kann. Der Inner-Pair-Effekt kann für Hauptgruppenmetalloxide mit (maximaler Oxidationsstufe - 2)
ebenfalls die Koordinationsgeometrie der O-Anionen um die M-Kationen
bestimmen.
Die Gesamtstruktur folgt
aus der Art der Verknüpfung der MOn-Polyeder.
Die Verknüpfung sollte dabei in der Weise erfolgen, daß gleichnamige
Kontakte maximiert und ungleichnamige minimiert werden.
Für die Art der Polyederverknüpfung gilt die
3. Pauling-Regel :
Gemeinsame Kanten und besonders gemeinsame Flächen destabilisieren eine
Struktur. Dies gilt insbesondere für hochgeladene Kationen mit kleiner
Koordinationszahl.
Bei den verschiedenen Modifikationen des TiO2
läßt sich die Auswirkung dieser Regel direkt erkennen:
Die Rutil-Modifikation (Verknüpfung über zwei gemeinsame Kanten
ist stabiler als Brookit (drei gemeinsame Kanten), dieser ist wiederum
stabiler als Anatas (vier gemeinsame Kanten).
Auf der Seite zu dichten Packungen findet sich
eine Struktursystematik von Ionenkristallen
auf der Basis gefüllter Lücken in dichtesten Packungen der
Anionen, in die auch die meisten Oxide einbezogen werden können.
(S. hierzu auch die Vorlagen mit den Schemazeichnungen).
Zusätzliche Anmerkungen:
- Generell (nicht nur bei Oxiden!) ist die Besetzung von Tetraederlücken mit gemeinsamen Flächen
nicht möglich.
- Oxide mit besetzten Oktaederlücken mit gemeinsamen Flächen werden
nur bei sehr kleinen Ionen (z.B. im Korund) und nie bei MO-Verbindungen (kein NiAs-Typ)
realisiert.
- Bei Oxiden erfolgt die Besetzung von Tetraederlücken generell nur durch sehr kleine
Ionen (Li,Be).
- Fast alle Oxide MO bilden NaCl-Struktur (12 gemeinsame Kanten).
- Oxide MO2 bilden keine Schichtstrukturen wie
CdCl2 oder CdI2 (6 gemeinsame Kanten), sondern
bevorzugen Strukturtypen mit weniger gemeinsamen Kanten; z.B. ist der Rutil-Typ
(2 gemeinsame Kanten) sehr häufig.
Im folgenden werden für alle Metalloxide (geordnet nach Zusammensetzung)
die Strukturen diskutiert, wobei auf die unter 3.3.1 (z.B. LFSE)
und 3.3.2 (Packungen) genannten Punkte jeweils hingewiesen wird.
Von der Diskussion
ausgenommen sind die Suboxide und Oxide mit O-O-Bindungen sowie
nichtstöchiometrische und z.T. auch gemischtvalente Oxide.